| Issue |
Biologie Aujourd'hui
Volume 211, Number 3, 2017
|
|
|---|---|---|
| Page(s) | 223 - 228 | |
| Section | La biologie computationnelle parle à la biologie expérimentale | |
| DOI | https://doi.org/10.1051/jbio/2017031 | |
| Published online | 7 février 2018 | |
Article
Bricoler avec les réseaux d’interactions protéines-protéines, leurs structures et leurs mutations associées aux maladies
Protein-protein interacting networks, their structures and disease-related mutations
Randall Division of Cellular and Molecular Biology, King’s College,
London, UK
* Auteur correspondant : Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Reçu :
20
Septembre
2017
Résumé
Au cours des dernières années, les comparaisons d’interactomes protéiques ont mis en évidence des modules conservés qui pourraient représenter des noyaux fonctionnels communs d’origine ancestrale. Dans ce contexte, des analyses récentes des réseaux d’interactions protéines-protéines ont conduit à un débat sur l’influence de la méthode expérimentale sur la qualité et la pertinence biologique de ces données d’interaction. Il est crucial de savoir dans quelle mesure les divergences entre les réseaux d’espèces différentes reflètent les biais d’échantillonnage des méthodes expérimentales respectives, par opposition aux caractéristiques topologiques dues à la fonctionnalité biologique. Cela nécessite des outils mathématiques nouveaux, précis et pratiques pour quantifier et comparer les structures topologiques des réseaux à haute résolution. À cette fin, nous avons étudié la relation entre les ensembles de graphes aléatoires structurés et les réseaux de signalisation biologiques réels, en mettant l’accent sur le nombre de cycles de graphes dans les réseaux, qui représentent des complexes dans les interactomes protéiques expérimentaux. En combinant des méthodes pour la dynamique des graphes et des algorithmes pour le comptage de boucles, nous estimons l’importance relative des boucles dans les réseaux biologiques par rapport aux analyses des réseaux.
Abstract
In recent years, the comparison of protein interactomes has identified conserved modules, that could represent functional nuclei with a common ancestry. Within this context, recent analyses of protein-protein interacting networks have led to a debate on the influence of the experimental method on the quality and biological pertinence of these data. It is crucial to understand the measure in which divergence between networks of different species reflect sampling biases in respective experimental methods, as opposed to topological features dictated by biological functionality. This aspect requires novel, precise and practical mathematical tools, to quantify and compare high resolution networks. To this end, we have studied the relationship between pools of aleatory graphs and real biological signalization networks, while stressing the number of graph cycles in the networks, which represent complexes in experimental protein interactomes. By combining methods for graph and algorithm dynamics to count the loops, we evaluate the relative importance of the loops in biological networks in comparison with network analyses.
Mots clés : Réseaux d’interactions protéiques / analyse structurelle et fonctionnelle du polymorphisme mononucléotidique / structure tridimensionnelle des complexes des protéines / théorie des graphiques aléatoires
Key words: protein-protein interacting networks / protein interactomes / structured aleatory graphs
© Société de Biologie, 2018
Abréviations
EM : Microscopie électronique
IC : Isoforme complète
IPP : Interactions protéines–protéines
PDB : Protein Data Bank
nsSVN : non-synonym Single Nucleotide Variant
SNP : Single Nucleotide Polymorphism
UniPPIN : Réseau IPP unifié
Contexte
La révolution génomique des dernières années a stimulé l’apparition de nouvelles technologies et de nouveaux outils d’analyse efficaces et précis pour la cartographie rapide et précise des gènes dans les génomes de milliers d’espèces (Koepfli et al., 2015). Ces progrès ont permis de fournir un atlas des gènes humains relativement exhaustif (Telenti et al., 2016). Cependant, cette information précieuse demeure insuffisante pour comprendre les relations fonctionnelles entre ces gènes et leurs répercussions à l’échelle de la cellule. Les produits des gènes, les protéines, jouent un rôle essentiel dans le bon fonctionnement de la cellule en constituant un réseau de communication au sein de la cellule par des interactions entre elles mais aussi avec les gènes.
Ainsi, caractériser et comprendre le fonctionnement des interactions protéines–protéines est essentiel pour améliorer notre compréhension des réseaux de communication existant dans la cellule. Par conséquent, un des nouveaux défis de la biologie réside dans l’identification exhaustive des interactions protéines–protéines (IPP) et la caractérisation des mécanismes moléculaires régissant ces interactions fonctionnelles.
Malheureusement, la cartographie de l’espace multidimensionnel d’interactions qui, par définition, résultent de processus dynamiques est beaucoup plus difficile à réaliser que l’annotation de génomes. De nombreuses techniques très différentes en complexité et efficacité ont ainsi été développées pour répondre à ce problème.
Récemment, plusieurs études réalisées sur différents protéomes ont été menées afin de comparer les données d’interactions protéines–protéines produites à l’échelle systémique par différentes approches (réseaux d’interactions construits à partir d’expériences de double hybride, de TAP (Tandem Affinity Purification) ou de spectrométrie de masse) ainsi que des données de structures 3D (Mosca et al., 2013 ; Lu et al., 2016). Malgré les progrès réalisés grâce à ces méthodes, le recouvrement des données recueillies par les différentes techniques demeure encore faible.
Une façon de tirer profit de ces informations ou instantanés expérimentaux est de les intégrer en les stockant, les cataloguant et les assemblant et ainsi obtenir une vision kaléidoscopique des IPP.
À partir de ces informations partielles ou instantanées, on peut émettre des hypothèses sur les réseaux que formeraient des sous-ensembles de protéines unis en modules fonctionnels. L’avantage de se focaliser sur des sous-ensembles topologiquement liés, annotés et bien décrits est d’affecter des scores de confiance aux unités modulaires fonctionnelles prédites par ces expériences (Havugimana et al., 2012 ; Chung et al., 2015). Sur le plan informatique, l’écart entre la connaissance des gènes et celle du réseau de communication de la cellule au travers des réseaux d’interactions protéiques peut être comblé par des cartes d’IPP prédites in silico où les interactions peuvent être pondérées en fonction de leur fiabilité et de leur annotation. L’intérêt est que la prédiction in silico est plus rapide et moins coûteuse. Par ailleurs, les résultats de ces prédictions informatiques peuvent être validés par des expériences ciblées permettant une utilisation plus efficace des ressources expérimentales (Carlin et al., 2011).
Nous analyserons aujourd’hui l’importance de l’extraction de modules topologiques récurrents dans les réseaux d’interactions protéiques en relation avec des mutations pathogènes affectant les protéines constituantes des modules. La comparaison de ces données dans le contexte de différentes pathologies devrait aider à mettre en évidence les modules essentiels qui pourraient servir de cibles pour la conception de nouvelles stratégies thérapeutiques.
Bricoler les réseaux d’interactions protéines–protéines
Construction d’un ensemble d’interactions protéines–protéines humaines
Récemment, nous avons intégré un ensemble de données provenant de neuf sources différentes d’interactions protéines–protéines humaines incluant des méga-bases de données et des données provenant d’études récentes réalisées à grande échelle. Lors de l’intégration, nous avons utilisé les numéros d’accès Uniprot (collectés le 15 mars 2017) pour fusionner différents formats de jeux de données et générer un réseau IPP unifié (UniPPIN) non redondant. Les détails des sources sont décrits dans le tableau et la figure 1.
 |
Figure 1 Assemblage de la Database UniPPIN et méthodes de détermination choisies. La base de données est disponible sur demande aux auteurs. |
Communauté de protéines
Nous avons développé une méthode permettant d’extraire des protéines à partir d’un réseau d’intérêt en recherchant des sous-réseaux formant des motifs particuliers (i.e. petits cycles) (Chung et al., 2015).
Nous avons ensuite appliqué cette méthode pour analyser les sous-réseaux IPP contenant des mutations et l’UniPPIN.
En combinant des méthodes de dynamique des graphes et des algorithmes de dénombrement de cycles, nous pouvons estimer l’importance relative des cycles observés dans les réseaux biologiques par rapport au nombre de cycles attendus dans des graphes aléatoires. Nous montrons que les cycles sont une caractéristique prédominante des IPP, suggérant que l’enrichissement de leur occurrence a un rôle fonctionnel clé. Nous démontrons que ces cycles contiennent des informations pertinentes sur les mécanismes biologiques sous-jacents au fonctionnement de la cellule (Figure 2). Leur étude peut ainsi permettre l’identification de modules essentiels associés à des fonctions critiques. De plus, ces modules hébergent généralement des fonctions reliées biologiquement et leur analyse peut ainsi permettre :
-
de compléter et valider l’annotation fonctionnelle des protéines associées à ces modules ;
-
d’étendre les annotations découlant de l’analyse des voies biologiques, comme cela a été le cas pour les protéines du cycle cellulaire.
Enfin, ces modules peuvent être utilisés pour concevoir des analyses expérimentales ciblées pour valider les prédictions ou identifier de nouvelles protéines associées à des complexes anormaux dans des maladies spécifiques.
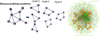 |
Figure 2 Définition des cycles courts. Dans un réseau d’interactions protéines–protéines (réseau en vert), les protéines (A, B, C, D, E, F, G, I) sont représentées dans les nœuds (p.e. A:B, A:C, B:C, B:G). Les liens n’ont pas d’information directionnelle indiquant leurs interactions mutuelles. Des boucles courtes de longueurs 3, 4, 5 sont représentées dans ce réseau. |
Outils Web pour la cartographie des mutations liées aux maladies
Dans le but de démontrer l’importance de connaître la structure en 3D des protéines, nous avons développé des outils pour l’analyse rapide et précise de l’impact de la variation génétique sur leurs domaines composants. On peut considérer les protéines en tant que domaines isolés ou complexes binaires extraits des réseaux.
PinSnps : analyse structurelle et fonctionnelle des SNP dans le cadre des réseaux d’interactions protéiques
PinSnps (Lu et al., 2016) est un pipeline de calcul pour effectuer facilement des analyses de données de réseaux d’interactions protéines–protéines. Par ailleurs, nous livrons aussi une prédiction de l’impact des mutations sur la stabilité d’une protéine d’intérêt en procurant à l’utilisateur les scores obtenus par un ensemble de méthodes différentes. Nous avons cartographié au total 2587 SNP liés à des maladies génétiques provenant de OMIM, 587.873 variants liés au cancer provenant de COSMIC et 1.484.045 SNP issus de dbSNP. Toutes ces données peuvent être téléchargées par l’utilisateur avec un R-script pour calculer l’enrichissement des SNP/variants dans les régions structurelles sélectionnées.
PinSnps est disponible à l’adresse http://fraternalilab.kcl.ac.uk/PinSnps/.
TITINdb
TITINdb : application web dédiée aux annotations relatives à la protéine titine, intégrant des informations de structure, de séquence, d’isoformes, de variants génétiques ainsi que des renseignements en lien avec les maladies qui lui sont associées (Lopes et al., 2013 ; Chauveau et al., 2014 ; Laddach et al., 2017).
La titine est la plus grande protéine connue, chaque molécule de titine recouvrant la moitié d’un sarcomère musculaire. L’isoforme complète (IC) la plus longue est constituée de 35 991 acides aminés (Figure 3). La protéine présente une structure modulaire composée principalement de domaines Ig et Fn3 et d’un seul domaine kinase. Dans TITINdb, les utilisateurs peuvent sélectionner des structures PDB (Protein Data Bank) pour visualiser les domaines dont la structure 3D est disponible. Récemment, le gène codant pour la titine est apparu comme un gène d’intérêt car ses variants sont associés à différentes myopathies telles que HCM (Hypertrophic Cardiomyopathy), DCM (Dilated Cardiomyopathy), HMERF (Hereditary Myopathy with Early Respiratory Failure) (Lopes et al., 2013 ; Chauveau et al., 2014).
Malheureusement, en raison de la grande taille de la protéine, la majorité des individus sans pathologie évidente possède un ou plusieurs variants rares de titine (Lopes et al., 2013). Cela aboutit au paradoxe que ces variants rares sont communément trouvés ; par conséquent la pathogénicité, dans ce cas, ne peut être déduite de la fréquence seule.
TITINdb a permis de visualiser les nsSVN associés aux maladies, provenant du projet 1000 génomes sur la structure 3D des différents domaines de la titine. De plus, l’impact de ces nsSNV sur la stabilité des domaines a été prédit par des méthodes in silico reposant sur des indications de structure et/ou de séquence.
Un schéma de numérotation cohérent pour les domaines et positions de résidus de la titine a été mis en place afin de cartographier toutes les positions d’intérêt sur l’isoforme IC de référence. Les utilisateurs peuvent facilement passer d’une isoforme à l’autre lors d’une recherche par position. Les limites des différents domaines de la titine ont également été définies en intégrant l’information de séquence et des résultats expérimentaux.
Lorsqu’aucune indication expérimentale de structure (par Radiographie X ou Résonance Magnétique Nucléaire) n’est disponible, des modèles d’homologie sont fournis. De plus, les utilisateurs peuvent télécharger les structures de domaines qu’eux-mêmes ont obtenues pour cartographier les nsSNV.
TITINdb est disponible à l’adresse : http://fraternalilab.kcl.ac.uk/TITINdb/.
 |
Figure 3 A) Organigramme TITINdb avec les méthodes utilisées. B) Les nsSNV associés à HMERF et TMD sont affichés. Les utilisateurs peuvent identifier les nsSNV associés à la maladie à partir du tableau SNV sur la structure du domaine et les visualiser par rapport à la distribution des nsSNV de la population (gnomAD ou 1000 génomes). Comme on peut le voir, ces deux groupes sont très distincts quand on les visualise sur la structure tridimensionnelle du domaine. Les analyses in silico pré-calculées sont présentées dans le tableau SNV. |
Perspectives
Dans cette communication, nous nous sommes concentrée sur l’extraction et la caractérisation des complexes d’interactions binaires obtenus par différentes approches expérimentales, par inférence et par prédiction. Cependant, la plupart des complexes biologiques qui exercent un rôle fonctionnel crucial dans la cellule sont constitués de multiples composants protéiques. Les méthodes basées sur la modélisation peuvent être utilisées pour représenter de tels assemblages, mais elles sont limitées par l’absence de structures établies pour certains composants. La caractérisation structurale de tels assemblages demeure encore aujourd’hui particulièrement difficile à résoudre même par les techniques modernes. Cependant, les progrès récents en cryo-EM permettent aujourd’hui la caractérisation structurale de certains grands complexes à une résolution proche de la résolution atomique (Lengyel et al., 2014 ; Chlanda & Krijnse Locker, 2017 ; Orlov et al., 2017). De plus, cette technologie peut être complétée par des données provenant d’expériences de cristallographie et de RMN, car les structures de complexes plus petits ou de composants seuls peuvent être projetées sur des cartes de densité EM et aider à une reconstruction précise du complexe. Ainsi, l’organisation de la PDB a lancé un schéma pour le dépôt de modèles hybrides résultant d’une combinaison de techniques expérimentales (Burley et al., 2017), ce qui permettra une utilisation efficace des données intégrées dans les méthodes de prédiction.
Un des défis consistera à développer des prédicteurs en utilisant efficacement les informations multimériques disponibles et le niveau de résolution associé. Les développeurs de Swiss Model ont commencé à répondre à cette requête avec la sortie d’un nouveau pipeline permettant la modélisation automatisée de complexes oligomériques (Bertoni et al., 2017). En parallèle, plusieurs programmes commencent à traiter l’assemblage automatisé de plusieurs composants, bien que les procédures reposent encore principalement sur l’amarrage de paires de tous les composants complexes (Soni & Madhusudhan, 2017).
Tout au long de ce chapitre, nous avons vu comment les informations ponctuelles fournissant en instantané des IPP dans un temps et un contexte donnés peuvent être combinées et intégrées à l’aide de méthodes informatiques pour fournir une vision kaléidoscopique de l’univers des interactions protéiques. Cependant, ce n’est qu’à travers le développement et l’intégration continus de méthodes expérimentales et computationnelles que l’on pourra concevoir un jour une carte multidimensionnelle, complète et précise du paysage des interactions protéiques.
Remerciements
FF remercie Anne Lopes pour les discussions, suggestions et corrections à propos de ce texte.
Références
- Bertoni, M., Kiefer, F., Biasini, M., Bordoli, L., Schwede, T. (2017). Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci Rep, 7, 10480. [CrossRef] [PubMed] [Google Scholar]
- Burley, S.K., Kurisu, G., Markley, J.L., Nakamura, H., Velankar, S., Berman, H.M., Sali, A., Schwede, T., Trewhella, J. (2017). PDB-Dev: a prototype system for depositing integrative/hybrid structural models. Structure, 25, 1317-1318. [CrossRef] [PubMed] [Google Scholar]
- Carlin L.M., Evans, R., Milewicz, H., Fernandes, L., Matthews, D.R., Perani, M., Levitt, J., Keppler, M.D., Monypenny, J., Coolen, T., Barber, P.R., Vojnovic, B., Suhling, K., Fraternali, F., Ameer-Beg, S., Parker, P.J., Thomas, N.S., Ng, T. (2011). A targeted siRNA screen identifies regulators of Cdc42 activity at the natural killer cell immunological synapse. Sci Signal, 4, 201. [CrossRef] [Google Scholar]
- Chauveau, C., Rowell, J., Ferreiro, A. (2014). A rising titan: TTN review and mutation update. Hum Mutat, 35, 1046-1059. [CrossRef] [PubMed] [Google Scholar]
- Chlanda, P., Krijnse Locker, J. (2017). The sleeping beauty kissed awake: new methods in electron microscopy to study cellular membranes. Biochem J, 474, 1041-1053. [CrossRef] [PubMed] [Google Scholar]
- Chung, S.S., Pandini, A., Annibale, A., Coolen, A.C., Thomas, N.S., Fraternali, F. (2015). Bridging topological and functional information in protein interaction networks by short loops profiling. Sci Rep, 5, 8540. [CrossRef] [PubMed] [Google Scholar]
- Havugimana, P.C., Hart, G.T., Nepusz, T., Yang, H., Turinsky, A.L., Li, Z., Wang, P.I., Boutz, D.R., Fong, V., Phanse, S., Babu, M., Craig, S.A., Hu, P., Wan, C., Vlasblom, J., Dar, V.U., Bezginov, A., Clark, G.W., Wu, G.C., Wodak, S.J., Tillier, E.R., Paccanaro, A., Marcotte, E.M., Emili, A. (2012). A Census of Human Soluble Protein Complexes. Cell, 150, 1068-1081. [CrossRef] [PubMed] [Google Scholar]
- Koepfli, K.P., Paten, B. Genome, 10K Community of Scientists, O’Brien, S.J. (2015). The Genome 10K Project: a way forward. Annu Rev Anim Biosci, 3, 57-111. [CrossRef] [PubMed] [Google Scholar]
- Laddach, A., Gautel, M., Fraternali, F. (2017). TITINdb—a computational tool to assess titin’s role as a disease gene. Bioinformatics, btx424. DOI: 10.1093/bioinformatics/btx424. [Google Scholar]
- Lengyel, J, Hnath, E., Storms, M., Wohlfarth, T. (2014). Towards an integrative structural biology approach: combining Cryo-TEM, X-ray crystallography, and NMR. J Struct Funct Genomics, 15, 117-124. [CrossRef] [PubMed] [Google Scholar]
- Lopes, L., Zekavati A., Syrris, P., Hubank, M., Giambartolomei, C., Dalageorgou, C., Jenkins, S., McKenna, W., UK 10K Consortium, Plagnol, V., Elliott, P.M. (2013). Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J Med Genet, 50, 228-239. [CrossRef] [PubMed] [Google Scholar]
- Lu, H.C., Herrera Braga, J., Fraternali, F. (2016). Pinsnps: structural and functional analysis of snps in the context of protein interaction networks. Bioinformatics, 32, 2534. [CrossRef] [PubMed] [Google Scholar]
- Mosca, R., Ceol, A., Aloy, P. (2013). Interactome3D: adding structural details to protein networks. Nat Methods, 10, 47-53. [CrossRef] [PubMed] [Google Scholar]
- Orlov, I., Myasnikov, A.G., Andronov, L., Natchiar, S.K., Khatter, H., Beinsteiner, B., Ménétret, J.F., Hazemann, I., Mohideen, K., Tazibt, K., Tabaroni, R., Kratzat, H., Djabeur, N., Bruxelles, T., Raivoniaina, F., Pompeo, L.D., Torchy, M., Billas, I., Urzhumtsev, A., Klaholz, B.P. (2017). The integrative role of cryo electron microscopy in molecular and cellular structural biology. Biol Cell, 109, 81-93. [CrossRef] [PubMed] [Google Scholar]
- Soni, N., Madhusudhan, M.S. (2017). Computational modeling of protein assemblies. Curr Opin Struct Biol, 44, 179-189. [CrossRef] [PubMed] [Google Scholar]
- Telenti, A., Pierce, L.C., Biggs, W.H., di Iulio, J., Wong, E.H., Fabani, M.M., Kirkness, E.F., Moustafa, A., Shah, N., Xie, C., Brewerton, S.C., Bulsara, N., Garner, C., Metzker, G., Sandoval, E., Perkins, B.A., Och, F.J., Turpaz, Y., Venter, J.C. (2016). Deep sequencing of 10,000 human genomes. Proc Natl Acad Sci USA, 113, 11901-11906. [CrossRef] [Google Scholar]
Citation de l’article : Fraternali, F. (2017). Bricoler avec les réseaux d’interactions protéines-protéines, leurs structures et leurs mutations associées aux maladies. Biologie Aujourd'hui, 211, 223-228
Liste des figures
|
Figure 1 Assemblage de la Database UniPPIN et méthodes de détermination choisies. La base de données est disponible sur demande aux auteurs. |
| Dans le texte | |
|
Figure 2 Définition des cycles courts. Dans un réseau d’interactions protéines–protéines (réseau en vert), les protéines (A, B, C, D, E, F, G, I) sont représentées dans les nœuds (p.e. A:B, A:C, B:C, B:G). Les liens n’ont pas d’information directionnelle indiquant leurs interactions mutuelles. Des boucles courtes de longueurs 3, 4, 5 sont représentées dans ce réseau. |
| Dans le texte | |
|
Figure 3 A) Organigramme TITINdb avec les méthodes utilisées. B) Les nsSNV associés à HMERF et TMD sont affichés. Les utilisateurs peuvent identifier les nsSNV associés à la maladie à partir du tableau SNV sur la structure du domaine et les visualiser par rapport à la distribution des nsSNV de la population (gnomAD ou 1000 génomes). Comme on peut le voir, ces deux groupes sont très distincts quand on les visualise sur la structure tridimensionnelle du domaine. Les analyses in silico pré-calculées sont présentées dans le tableau SNV. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.