| Numéro |
Biologie Aujourd’hui
Volume 216, Numéro 3-4, 2022
|
|
|---|---|---|
| Page(s) | 125 - 130 | |
| DOI | https://doi.org/10.1051/jbio/2022013 | |
| Publié en ligne | 6 février 2023 | |
Article
Approches moléculaires et thérapeutiques des interactions entre l’ocytocine et son récepteur
Oxytocin and its receptor: molecular and therapeutic approaches
Laboratoire d’Innovation Thérapeutique, UMR7200, Faculté de Pharmacie de Strasbourg, 74 route du Rhin, 67400 Illkirch, France
* Auteur correspondant : mhibert@unistra.fr
Reçu :
27
Juin
2022
L’ocytocine est une neurohormone connue à l’origine pour son rôle dans les contractions de l’utérus au moment de l’accouchement et les contractions des glandes mammaires pour permettre l’éjection du lait lors de la tétée. Depuis les 25 dernières années, de multiples autres effets centraux et périphériques ont été identifiés, notamment dans les processus d’attachement entre parents et enfant, entre adultes et entre un individu et son groupe social. Nous avons abordé au cours de cette période la question fondamentale de l’architecture structurale et fonctionnelle du complexe formé par l’ocytocine et son récepteur et l’application de ce savoir à la conception de candidats médicaments. Ceci a conduit à la découverte du premier agoniste non peptidique de l’ocytocine, le LIT-001, restaurant l’interaction sociale dans un modèle animal d’autisme.
Abstract
It is known since the fifties that oxytocin is a neurohormone synthesized in the brain and released in blood circulation to trigger uterus contraction during delivery. It is also involved in milk ejection during breast-feeding. Over the past 25 years, many other central and peripheral functions have been discovered, in particular for attachment between child and parents as well as between individuals and interaction between a human being and its social group. Over this period, we have studied the functional supramolecular architecture of the hormone bound to its receptor. This information was used to design pharmacological probes and drug candidates. This led to the discovery of the first non-peptide oxytocin receptor full agonist. This molecule, LIT-001, restores social interaction in an animal model of autism and paves the way for a treatment of this neurodevelopmental disorder.
Mots clés : ocytocine et son récepteur / interactions moléculaires / agoniste / autisme / candidat médicament
Key words: oxytocin and its receptor / molecular interactions / oxytocin agonist / autism / drug candidate
© Société de Biologie, 2023
Abréviations
FRET : Transfert d’énergie entre molécules fluorescentes
OTR : Récepteur de l’ocytocine
TSA : Troubles du spectre autistique
V1AR : Récepteur 1A de la vasopressine
Introduction
Les sciences du vivant abordent quelques questions essentielles : qu’est-ce que la vie ? Qu’est-ce que la mort ? Qu’est-ce que la maladie ? Quels en sont les mécanismes biologiques et moléculaires fondamentaux ? Il nous a semblé important d’aborder dès les années 1990 une question tout aussi importante à nos yeux : qu’est-ce que l’amour ? Cette énigme avait évidemment été largement abordée depuis des siècles par le biais des sciences humaines et sociales, par les artistes ainsi que par tout un chacun. Les sciences dites « dures » n’avaient cependant pas encore trouvé de porte d’entrée pour étudier ce comportement étrange et particulier, si important pour l’individu et nos sociétés. Elle leur fut ouverte principalement par le travail de collègues nord-américains, un zoologue, Lowell Lee Getz, et trois pharmacologues, Sue Carter, Thomas Insel et Larry Young.
La contribution de Lowell Lee Getz est quelque peu oubliée, elle est pourtant essentielle. Il a passé une bonne partie de sa vie de scientifique, des années 1970 à 1990, à observer des campagnols vivant dans divers environnements naturels aux États-Unis. Il a observé des différences très significatives entre les animaux des prairies et ceux vivant en milieu plus hostile, les montagnes. Les premiers forment des couples monogames et s’occupent, père comme mère, de leur progéniture avec attention. Les seconds sont par contre polygames et seule la mère élève ses petits le temps minimum nécessaire à leur sevrage. Ses collègues pharmacologues furent alertés à propos de ces comportements radicalement opposés au sein d’une même espèce animale, qu’ils confirmèrent en laboratoire (Carter & Getz, 1993). Des différences au niveau pharmacologique et génétique furent alors recherchées et découvertes : les deux groupes se distinguaient principalement par les taux de vasopressine (AVP) et d’ocytocine (OT) secrétées par leurs cerveaux et par les taux et les zones d’expression de leurs récepteurs respectifs V1AR et OTR. De manière spectaculaire, les comportements d’attachement des deux groupes purent être respectivement inversés par une administration d’OT et le blocage de son action (Williams et al., 1994). Ces résultats ouvrirent la voie à une exploration plus systématique des effets centraux de ces deux neurohormones et à l’évaluation de leur potentiel thérapeutique.
L’ocytocine et son récepteur, architecture moléculaire fonctionnelle
L’ocytocine est un nonapeptide cyclisé par un pont disulfure entre les cystéines en positions 1 et 6 (Figure 1). Malgré cette structure partiellement cyclique, la molécule peut adopter un très grand nombre de conformations différentes dans une faible fourchette d’énergie, ce qui rend difficile la prédiction de sa conformation active. Son partenaire supramoléculaire principal, le récepteur OTR, comporte 389 acides aminés. Il appartient à la classe génomique et structurale des récepteurs couplés aux protéines G qui comprend environ 800 membres. Leurs séquences ont commencé à être identifiées à partir des années 1985, par contre leur nature membranaire a considérablement compliqué l’accès à leur structure cristallographique. Face à cette difficulté, nous avons entrepris dès 1990 de proposer les premiers modèles moléculaires détaillés de cette famille de récepteurs, notamment ceux de la dopamine, de la noradrénaline, de la sérotonine et de l’acétylcholine (Hibert et al., 1991 ; Trumpp-Kallmeyer et al., 1992). Un site de liaison a pu être proposé pour chacun de ces neurotransmetteurs au sein des sept hélices transmembranaires. Ces modèles ont depuis été qualitativement validés par la résolution de leur structure cristallographique. Dans le cas particulier de l’AVP et de l’OT, le défi était plus relevé du fait de la taille et de la nature peptidique de ces neurohormones. Nous avons pu générer le modèle tridimensionnel des récepteurs V1AR et OTR et identifier au sein des structures une poche profonde parfaitement complémentaire des ligands endogènes (Hibert et al., 1999). Toutes les interactions entre les chaînes latérales des hormones et celles de la poche de leurs récepteurs respectifs ont été caractérisées. Les résidus hydrophobes 2 et 3 de l’OT et de l’AVP se logent dans la zone hydrophobe de leurs récepteurs au niveau du fond de la poche, contre les hélices 5, 6 et 7. Les résidus plus hydrophiles interagissent quant à eux avec la couronne d’acides aminés à l’entrée de la poche. Un modèle moléculaire, même autocohérent, n’ayant d’intérêt que s’il est validé par l’expérience (Hibert et al., 1993), nous avons alors entrepris de démontrer une à une les interactions entre les chaînes latérales des ligands et celles des récepteurs, par mutagenèse dirigée. Les mutations en alanine des acides aminés bordant le site de fixation potentiel du ligand ont permis d’identifier les zones responsables de l’affinité, de l’efficacité et de la sélectivité de l’interaction récepteur-ligand (Hibert et al., 1995 ; Mouillac et al., 1995). L’interprétation de résultats de mutagenèse restant sujette à discussion, nous avons complété cette analyse par des expériences de marquage covalent, soit par la méthode classique de marquage de photoaffinité (Phalipou et al., 1999), soit par une nouvelle stratégie, le marquage covalent dirigé (Tahtaoui et al., 2003). Deux points d’ancrage du neuropeptide ont ainsi pu être confirmés, ce qui valide sans ambiguïté son positionnement dans le site actif du récepteur. Nous disposions donc dès la fin des années 1990 d’un modèle de l’interaction OT-OTR aussi précis et validé qu’il pouvait l’être à l’époque. Il aura fallu attendre 30 ans de plus pour que les cristallographes réussissent à produire une structure expérimentale du récepteur OTR complexé à un antagoniste (Waltenspühl et al., 2020), et plus récemment à l’ocytocine elle-même (Meyerowitz et al., 2022). Les modèles moléculaires initiaux et la réalité expérimentale étant en bon accord, nous les avons utilisés pour rationaliser la conception de sondes pharmacologiques et de candidats médicaments.
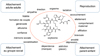 |
Figure 1 Fonctions modulées par l’ocytocine (adapté de Hibert, 2021). |
L’ocytocine et ses fonctions
L’OT a été initialement caractérisée pour ses actions sur la contraction de l’utérus au moment de l’accouchement et l’éjection du lait lors de l’allaitement du nouveau-né. Les travaux de Getz, Carter et Insel sur les campagnols ont ouvert la voie à de très nombreuses études sur d’autres fonctions centrales et périphériques de cette neurohormone que nous listerons ici, sans rentrer dans le détail, en renvoyant à deux ouvrages récents sur le sujet (Hibert, 2021 ; Freund-Mercier, 2022). Ces études ont été réalisées chez l’animal mais également pour nombre d’entre elles chez l’Homme, en utilisant principalement chez ce dernier la voie d’administration intranasale. En effet, l’OT est un gros peptide qui ne passe pas les barrières gastro-intestinale et centrale par administration orale, intradermique ou intraveineuse. Elle est par ailleurs peu stable dans le sang, sa durée de vie n’y étant que de quelques minutes seulement. L’utilisation chez l’humain d’un spray nasal permet le passage de faibles doses d’hormone vers le cerveau ce qui a rendu possible la démonstration d’un effet significatif de cette neurohormone dans quatre types de circonstances : la naissance, l’attachement parents-enfants, l’attachement entre individus et l’attachement d’un individu à son groupe social.
Dans les premiers mois de la grossesse, les taux d’OT produite par la mère semblent être un indicateur de la qualité du comportement maternel après la naissance. L’OT est nécessaire aux contractions musculaires de l’acte d’accouchement. C’est d’ailleurs cette neurohormone qui est injectée pour provoquer l’accouchement en cas de nécessité et pour limiter les risques post-partum. Elle semble par ailleurs pouvoir protéger le nouveau-né de la douleur et de l’hypoxie durant la naissance, limiter sa sensibilité à l’inflammation et réguler l’installation de son microbiote. Elle est nécessaire au bon neurodéveloppement de l’enfant et à l’établissement des liens affectifs avec les parents et à sa construction sociale. Elle limite par ailleurs les phases dépressives du post-partum des mères. L’OT permet l’apprentissage des règles d’interaction sociale, facilite l’empathie, accroît l’estime de soi et la confiance en l’autre. Lors de la naissance d’un sentiment amoureux, elle renforce l’attachement à l’être aimé et la mise à distance des autres. Elle est particulièrement secrétée lors de regards empathiques, de contacts peau à peau, de baisers et de l’orgasme, contribuant sans doute au renforcement de l’attachement. En règle générale, elle gomme les saillances, ramenant à la normale les comportements d’interaction sociale déviants. Son principal défaut, ou sa qualité selon le point de vue où l’on se place, est d’augmenter l’agressivité si le groupe auquel appartient la personne est menacé. Remarquablement conservée au cours de l’évolution, comme l’est également son unique récepteur, cette neurohormone représente un des acteurs clés de la survie des espèces animales. On peut même envisager de l’utiliser comme marqueur biologique, voire comme traitement de divers états pathologiques,
Agoniste de l’ocytocine pour traiter l’autisme
Des études chez l’animal ont montré qu’un rongeur privé d’activation ocytocinergique à la naissance ne développe pas d’interaction sociale. Cette observation a conduit à penser que l’OT pouvait jouer un rôle dans les troubles du spectre autistique (TSA). Cette hypothèse a rapidement été confirmée par l’observation de taux anormaux de cette neurohormone chez les patients et par le fait que des mutations du gène du récepteur OTR pouvaient dans quelques cas conduire à des TSA. L’existence d’un médicament à base d’OT sous forme de spray nasal (le SyntocinonR) a permis de montrer l’impact de l’administration de cette neurohormone sur plusieurs traits autistiques, tels que les stéréotypies (Hollander et al., 2003), les déficits de perception des émotions sur un regard ou un visage, les troubles de la communication et des interactions sociales (Andari et al., 2010), apportant ainsi la preuve de concept que l’activation du récepteur OTR ouvrait une perspective thérapeutique pour les TSA.
L’OT elle-même ne peut pas être développée comme traitement de l’autisme pour plusieurs raisons : son inactivité par voie orale, sa très courte durée de vie dans le corps, son manque de spécificité et l’impossibilité de la breveter pour cette application. Le défi pour les pharmacochimistes consiste alors à concevoir un agoniste spécifique du récepteur OTR, biodisponible, stable, brevetable et capable d’agir par voie orale sur des comportements relevant des TSA. Trois stratégies ont été explorées par notre laboratoire pour atteindre cet objectif : l’approche mécanistique, le criblage et l’optimisation des relations structure-efficacité.
La première s’appuie sur les modèles moléculaires tridimensionnels de l’OT en interaction avec son récepteur. Nous avions montré que les résidus Tyr-Ile-Gln de l’OT en interaction avec les hélices 5, 6 et 7 d’OTR étaient cruciaux pour induire l’agonisme (Chini et al., 1997). Partant de ce résultat, nous avons entrepris de concevoir des petites molécules non peptidiques mimant ces trois acides aminés dans la conformation modélisée. Parmi les molécules conçues et synthétisées, une aza-dicétopipérazine a effectivement montré une affinité remarquable de 0,8 micromolaire pour le récepteur. Elle s’est malheureusement révélée être un antagoniste de l’OTR, de même que les autres analogues testés.
La deuxième stratégie que nous avons mise en œuvre en vue de disposer d’un agoniste consiste à avoir recours au criblage à moyen ou haut débit. Un essai de liaison a été mis au point et robotisé. Il repose sur la technologie de transfert d’énergie entre molécules fluorescentes (FRET). Pour ce faire, le récepteur OTR a été fusionné à une protéine fluorescente verte et un agoniste peptidique porteur du fluorophore Lissamine a été développé (Karpenko et al., 2015). La liaison entre les deux partenaires du complexe peut alors être suivie par FRET entre les deux entités. Le rapport signal sur bruit est excellent et permet de miniaturiser et robotiser l’essai. Environ 20 000 molécules extraites de la Chimiothèque Nationale (https://chembiofrance.cn.cnrs.fr/fr/composante/chimiotheque) ont ainsi été testées. Quelques « touches micromolaires » ont été identifiées, mais toutes se sont avérées être des antagonistes du récepteur dans les tests fonctionnels et non pas les agonistes recherchés.
La troisième stratégie a consisté à modifier systématiquement les structures d’antagonistes et d’un agoniste partiel connus pour obtenir l’agoniste complet attendu. Il existe en effet plusieurs antagonistes non peptidiques de l’OTR mais le travail autour de leur structure n’a pas apporté d’agonisme. Par contre, un agoniste partiel avait été décrit marginalement dans un article de la société Ferring (Figure 2 ; Pitt et al., 2004). Ce composé présente une relativement faible affinité et sélectivité pour le récepteur OTR, et une faible efficacité comparée à l’OT (Tableau 1). Un analogue de la société Wyeth a ensuite été publié (Rahman et al., 2007). Son affinité pour OTR est meilleure mais sa sélectivité et son efficacité restent insuffisantes (Hicks et al., 2015). Nous avons modifié systématiquement ces structures, ce qui a conduit dans la plupart des cas à une perte d’affinité ou d’efficacité fonctionnelle (Frantz et al., 2010). Un seul composé dans la série a montré une pleine activité d’agoniste avec un profil de sélectivité amélioré, il s’agit de la molécule LIT-001. En effet, ce composé présente une affinité de 61 nM pour OTR, une relative sélectivité vis-à-vis du récepteur V1AR de l’AVT, et pour la première fois une efficacité totale sur un test fonctionnel cellulaire. Il est de nature non peptidique, stable chimiquement, de masse moléculaire faible comparée à celle de l’OT (555 au lieu de 1000), avec une hydrophilie et une polarité compatibles avec le passage dans le cerveau. Ces caractéristiques en font un bon candidat pour l’étude de son efficacité dans des modèles animaux des TSA.
 |
Figure 2 Structures des agonistes partiels non peptidiques de l’ocytocine et de l’agoniste plein LIT-001. |
Affinité et efficacité des agonistes partiels et complets non peptidiques du récepteur OTR.
LIT-001 et autisme
Les TSA sont de génotypes et de phénotypes très divers. Il semble difficile de réaliser des modèles animaux incontestablement représentatifs de la pathologie chez l’humain. On peut cependant s’en rapprocher au mieux et, de ce point de vue, le modèle développé par Jérôme Becker et Julie Le Merrer nous a semblé pertinent (Becker et al., 2014). Ils ont notamment montré que les souris KO pour le récepteur mu de la morphine (gène Oprm1) présentent tous les symptômes primaires de la pathologie, y compris en termes de réponse pharmacologique. On retrouve un déficit de l’interaction sociale faiblement sensible au baclofène, des troubles de la communication sociale, des stéréotypies locomotrices et cognitives sensibles à la rispéridone. Tous les troubles secondaires sont également présents : anxiété, agressivité, locomotion, seuil épileptique abaissé, troubles gastro-intestinaux, etc. Par ailleurs, l’administration d’OT a un effet bénéfique sur l’ensemble de ces symptômes. Ajoutons enfin que certains patients autistes ont des mutations sur le gène Oprm1. Le composé LIT-001 a été testé par administration intrapéritonéale à la dose de 10 mg/kg. Divers paramètres montrent son efficacité (Frantz et al., 2018). On observe par exemple que les souris contrôles passent 80 % de leur temps en interactions sociales alors que les souris Oprm1-KO n’y engagent que 17 % de leur temps environ. Après administration du LIT-001, rien ne change pour les souris sauvages mais les interactions sociales reviennent à la normale pour les souris KO traitées (Figure 3).
Il apparaît donc que le LIT-001 représente le premier agoniste du récepteur OTR montrant une activité significative dans un modèle animal d’autisme après administration périphérique. Des développements plus récents ont conduit à une molécule optimisée et brevetable active à des concentrations sub-nanomolaires avec une sélectivité encore améliorée, ouvrant la voie à un développement préclinique.
On peut envisager l’utilisation thérapeutique d’un agoniste ocytocinergique dans de nombreuses autres pathologies telles que la douleur, la dépression, certaines formes d’anxiété, les addictions aux drogues, les troubles de l’interaction sociale, ceux du comportement alimentaire, etc. De fait, le LIT-001 a d’ores et déjà démontré son efficacité dans le traitement de la douleur inflammatoire (Hilfiger et al., 2020). Des données non publiées indiquent également son efficacité dans le sevrage de l’alcool. D’autres données devraient suivre avec les nouvelles molécules encore plus actives.
 |
Figure 3 À la dose de 10 mg/kg par voie intrapéritonéale, le composé LIT-001 rétablit un temps de contact nasal normal chez les souris KO pour le récepteur opioïde mu (Oprm1-/-), modèle animal de TSA (d’après Frantz et al., 2018). |
Conclusion
L’OT est passée en vingt ans d’une « simple » implication dans la parturition et la lactation à une multitude de fonctions biologiques, physiologiques et sociales du plus grand intérêt. Bien évidemment non réductible à l’hormone de l’amour ou de l’attachement, elle contribue à un cercle d’actions toutes nécessaires à la survie de l’espèce, aussi bien chez l’animal que chez l’Homme. Cette exploration biologique et fonctionnelle doit être poursuivie avec des outils génétiques et pharmacologiques encore plus spécifiques, notamment pour discriminer l’activation des récepteurs V1AR de celle d’OTR. La communauté scientifique bute encore sur le développement de sondes d’imagerie in vivo permettant par exemple les études chez l’Homme par tomographie d’émission de positons. Au niveau du récepteur, l’impact de nombreux variants génétiques et épigénétiques est de plus en plus étudié et montre l’existence de prédispositions comportementales, pathologiques ou non, associées à ces mutations (Danoff et al., 2021).
Du point de vue thérapeutique, une première molécule, le LIT-001, ouvre de nouvelles perspectives pour le traitement des TSA, voire d’autres pathologies telles que les douleurs ou les addictions aux drogues. La publication récente de la structure cristallographique de l’OT fixée à son récepteur OTR devrait faciliter la découverte d’autres agonistes drug-like. Il faut cependant rappeler que l’OT à fortes doses peut induire une désensibilisation de son récepteur. Il y a par ailleurs des stades de développement ou d’interaction sociale au cours desquels elle joue un rôle particulièrement crucial (attachement, apprentissage, interactions empathiques), et toute action pharmacologique à ces stades pourrait se révéler critique. Enfin, certains de ses effets physiologiques pourraient peut-être passer par le récepteur V1AR plutôt que par le récepteur OTR. Il conviendra donc d’explorer l’intérêt thérapeutique d’agonistes tels que le LIT-001 avec discernement, pour le bénéfice optimal des patients.
Références
- Andari, E., Duhamel, J.-R., Zalla, T., Herbrecht, E., Leboyer, M., Sirigu, A. (2010). Promoting social behavior with oxytocin in high-functioning autism spectrum disorders. Proc Natl Acad Sci USA, 107, 4389-4394. [CrossRef] [PubMed] [Google Scholar]
- Becker, J.A., Clesse, D., Spiegelhalter, C., Schwab, Y., Le Merrer, J., Kieffer, B.L. (2014). Autistic-like syndrome in mu opioid receptor null mice is relieved by facilitated mGluR4 activity. Neuropsychopharmacology, 39, 2049-2060. [CrossRef] [PubMed] [Google Scholar]
- Carter, C.S., Getz, L.L. (1993). Monogamy and the prairie vole. Sci Am, 268, 100-106. [CrossRef] [PubMed] [Google Scholar]
- Chini, B., Mouillac, B., Ala, Y., Balestre, M.N., Trumpp-Kallmeyer, S., Hoflack, J., Elands, J., Hibert, M., Manning, M., Jard, S., Barberis, C. (1997). Identification of a single residue responsible for agonist selectivity in the Oxytocin–Vasopressin receptors. Ann NY Acad Sci, 812, 218-221. [CrossRef] [PubMed] [Google Scholar]
- Danoff, J.S., Wroblewski, K.L., Graves, A.J., Quinn, G.C., Perkeybile, A.M., Kenkel, W.M., Lillard, T.S., Parikh, H.I., Golino, H.F., Gregory, S.G., Carter, C.S., Bales, K.L., Connelly, J.J. (2021). Genetic, epigenetic, and environmental factors controlling oxytocin receptor gene expression. Clin Epigenetics, 13, 23. [CrossRef] [PubMed] [Google Scholar]
- Frantz, M.C., Rodrigo, J., Boudier, L., Durroux, T., Mouillac, B., Hibert, M. (2010). Subtlety of the structure-affinity and structure-efficacy relationships around a nonpeptide oxytocin receptor agonist. J Med Chem, 53, 1546-1562. [CrossRef] [PubMed] [Google Scholar]
- Frantz, M.C., Pellissier, L.P., Pflimlin, E., Loison, S., Gandía, J., Marsol, C., Durroux, T., Mouillac, B., Becker, J.A.J., Le Merrer, J., Valencia, C., Villa, P., Bonnet, D., Hibert, M. (2018). LIT-001, the first nonpeptide oxytocin receptor agonist that improves social interaction in a mouse model of autism. J Med Chem, 61, 8670-8692. [CrossRef] [PubMed] [Google Scholar]
- Freund-Mercier, M.J., Ocytocine : entre mythe et réalité, Doin Ed., Paris, 2022. [Google Scholar]
- Hibert, M., Trumpp-Kallmeyer, S., Bruinvels, A., Hoflack, J. (1991). Three-dimensional models of the cationic neurotransmitter G-protein coupled receptors. Mol Pharmacol, 40, 8-15. [PubMed] [Google Scholar]
- Hibert, M., Trumpp-Kallmeyer, S., Bruinvels, A., Hoflack, J. (1993). This is not a G protein-coupled receptor. Trends Pharmacol Sci, 14, 7-12. [CrossRef] [PubMed] [Google Scholar]
- Hibert, M., Hoflack, J., Trumpp-Kallmeyer, S., Paquet, J.L., Leppik, R., Barberis, C., Mouillac, B., Chini, B., Jard, S. (1995). 3D models of hormone receptors: experimental validation. Eur J Med Chem, 30, 189-199. [Google Scholar]
- Hibert, M., Hoflack, J., Trumpp-Kallmeyer, S., Mouillac, B., Chini, B., Mahé, E., Cotte, N., Jard, S., Manning, M., Barberis, C. (1999). Functional architecture of vasopressin/oxytocin receptors. J Recept Signal Transduct Res, 19, 589-596. [CrossRef] [PubMed] [Google Scholar]
- Hibert, M., Ocytocine mon amour, HumenSciences Ed., Paris, 2021. [Google Scholar]
- Hicks, C., Ramos, L., Reekie, T., Narlavar, R., Kassiou, M., McGregor, L. (2015). WAY 267,464, a non-peptide oxytocin receptor agonist, impairs social recognition memory in rats through a vasopressin 1A receptor antagonist action. Psychopharmacology, 232, 2659-2667. [CrossRef] [PubMed] [Google Scholar]
- Hilfiger, L., Zhao, Q., Kerspern, D., Inquimbert, P., Andry, V., Goumon, Y., Darbon, P., Hibert, M., Charlet, A. (2020). A nonpeptide oxytocin receptor agonist for a durable relief of inflammatory pain. Sci Rep, 10, 3017. [CrossRef] [PubMed] [Google Scholar]
- Hollander, E., Novotny, S., Hanratty, M., Yaffe, R., DeCaria, C.M., Aronowitz, B.R. (2003). Oxytocin infusion reduces repetitive behaviors in adults with autistic and Asperger’s disorders. Neuropsychopharmacology, 28, 193-198. [CrossRef] [PubMed] [Google Scholar]
- Karpenko, I.A., Margathe, J.F., Rodriguez, T., Pflimlin, E., Dupuis, E., Hibert, M., Durroux, T., Bonnet, D. (2015). Selective nonpeptidic fluorescent ligands for oxytocin receptor: design, synthesis, and application to time-resolved FRET binding assay. J Med Chem, 58, 2547-2552. [CrossRef] [PubMed] [Google Scholar]
- Meyerowitz, J., Robertson, M., Ximena Barros-Álvarez, X., Panova, O., Nwokonko, R., Gao, Y., Skiniotis, G. (2022). The oxytocin signaling complex reveals a molecular switch for cation dependence. Nat Struct Mol Biol, 29, 274-281. [CrossRef] [PubMed] [Google Scholar]
- Mouillac, B., Chini, B., Balestre, M.N., Jard, S., Barberis, C., Manning, M., Tribollet, E., Trumpp-Kallmeyer, S., Hoflack, J., Elands, J., Hibert, M. (1995). Identification of agonist binding sites of vasopressin and oxytocin receptors. Adv Exp Med Biol, 395, 301-310. [PubMed] [Google Scholar]
- Phalipou, S., Seyer, R., Cotte, N., Barberis, C., Hibert, M., Mouillac, B. (1999). Docking of linear peptide antagonists into the human V1a vasopressin receptor: identification of binding domains by photoaffinity. J Biol Chem, 274, 23316-23327. [CrossRef] [PubMed] [Google Scholar]
- Pitt, G.R., Batt, A.R., Haigh, R.M., Penson, A.M., Robson, P.A., Rooker, D.P., Tartar, A.L., Trim, J.E., Yea, C.M., Roe, M.B. (2004). Nonpeptide oxytocin agonists. Bioorg Med Chem Lett, 14, 4585-4589. [CrossRef] [PubMed] [Google Scholar]
- Rahman, Z., Resnick, L., Rosenzweig-Lipson, S.J., Ring, R.H. (2007). Methods of treatment using oxytocin receptor agonists. US patent application 2007/0117794 (published 2007-05-24). [Google Scholar]
- Tahtaoui, C., Balestre, M.N., Klotz, P., Rognan, D., Barberis, C., Mouillac, B., Hibert, M. (2003). Identification of the binding sites of the SR 49059 nonpeptide antagonist into the V1a vasopressin receptor using sulfydryl-reactive ligands and cysteine mutants as chemical sensors. J Biol Chem, 278, 40010-40019. [CrossRef] [PubMed] [Google Scholar]
- Trumpp-Kallmeyer, S., Bruinvels, J.A., Hibert, M. (1992) Modelling of G-protein coupled receptors: application to dopamine, serotonin, adrenaline, acetylcholine and mammalian opsin receptors. J Med Chem, 35, 3448-3462. [CrossRef] [PubMed] [Google Scholar]
- Waltenspühl, Y., Schöppe, J., Ehrenmann, J., Kummer, L., Plückthun, A. (2020). Crystal structure of the human oxytocin receptor. Sci Adv, 15, 6, eabb5419. [CrossRef] [PubMed] [Google Scholar]
- Williams, J.R., Insel, T.R., Harbaugh, C.R., Carter, C.S. (1994). Oxytocin administered centrally facilitates formation of a partner preference in female prairie voles (Microtus ochrogaster). J Neuroendocrinol, 6, 247-250. [CrossRef] [PubMed] [Google Scholar]
Citation de l’article : Hibert, M. (2022). Approches moléculaires et thérapeutiques des interactions entre l’ocytocine et son récepteur. Biologie Aujourd’hui, 216, 125-130
Liste des tableaux
Affinité et efficacité des agonistes partiels et complets non peptidiques du récepteur OTR.
Liste des figures
|
Figure 1 Fonctions modulées par l’ocytocine (adapté de Hibert, 2021). |
| Dans le texte | |
|
Figure 2 Structures des agonistes partiels non peptidiques de l’ocytocine et de l’agoniste plein LIT-001. |
| Dans le texte | |
|
Figure 3 À la dose de 10 mg/kg par voie intrapéritonéale, le composé LIT-001 rétablit un temps de contact nasal normal chez les souris KO pour le récepteur opioïde mu (Oprm1-/-), modèle animal de TSA (d’après Frantz et al., 2018). |
| Dans le texte | |
Les statistiques affichées correspondent au cumul d'une part des vues des résumés de l'article et d'autre part des vues et téléchargements de l'article plein-texte (PDF, Full-HTML, ePub... selon les formats disponibles) sur la platefome Vision4Press.
Les statistiques sont disponibles avec un délai de 48 à 96 heures et sont mises à jour quotidiennement en semaine.
Le chargement des statistiques peut être long.