| Issue |
Biologie Aujourd’hui
Volume 213, Number 1-2, 2019
|
|
|---|---|---|
| Page(s) | 51 - 57 | |
| DOI | https://doi.org/10.1051/jbio/2019020 | |
| Published online | 5 juillet 2019 | |
Article
Canaux potassiques à deux domaines P (K2P) et migraine
Two-pore-domain potassium channels and molecular mechanisms underlying migraine
1
Université Côte d’Azur, CNRS, INSERM, Institut de Biologie Valrose, Parc Valrose,
06108
Nice, France
2
Laboratories of Excellence, Ion Channel Science and Therapeutics,
Nice, France
* Auteur correspondant : Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Reçu :
4
Juin
2019
Résumé
La migraine est un désordre neurologique qui affecte 15 % de la population mondiale. Les crises migraineuses sont liées, entre autres, à l’hyperexcitabilité électrique des neurones trigéminaux. Leur activité électrique est contrôlée par les canaux potassiques à deux domaines P (K2P) dont l’importance dans l’induction du contrôle de l’excitabilité a récemment été mise en évidence par la découverte d’une version mutée de l’un d’eux TRESK, le mutant TRESK-MT, qui est lié à la migraine. Cette découverte a été controversée à la suite du séquençage d’autres canaux TRESK mutés non fonctionnels qui ne sont pas liés à la migraine. Notre étude montre que les mutations délétères du gène TRESK impliquées dans la migraine entraînent la formation de deux protéines au lieu d’une (comme attendu du gène non muté), via un nouveau mécanisme appelé fsATI (initiation alternative de la traduction induite par déplacement du cadre de lecture). L’une est inactive et l’autre, en ciblant d’autres canaux K2P, TREK1 et TREK2, stimule fortement l’activité électrique des neurones provoquant ainsi des crises migraineuses. Cette découverte a permis l’identification de deux nouvelles cibles dans le traitement de la migraine, TREK1 et TREK2, et suggère que les mutations induisant une traduction alternative due à un déplacement du cadre de lecture (fsATI) doivent être considérées comme une classe distincte de mutations lors de l’étude des mutations impliquées dans les pathologies humaines.
Abstract
Migraine is a common, disabling neurological disorder with genetic, environmental and hormonal components and a prevalence estimated at ∼15%. Migraine episodes are notably related, among several factors, to electric hyperexcitability in sensory neurons. Their electrical activity is controlled by ion channels that generate current, specifically by the two-pore-domain potassium, K2P, channels, which inhibit electrical activity. Mutation in the gene encoding TRESK, a K2P channel, causes the formation of TRESK-MT1, the expected non-functional C-terminal truncated TRESK channel, and an additional unexpected protein, TRESK-MT2, which corresponds to a non-functional N-terminal truncated TRESK channel, through a mechanism called frameshift mutation-induced Alternative Translation Initiation (fsATI). TRESK-MT1 is inactive but TRESK-M2 targets two other ion channels, TREK1 and TREK2, inducing a great stimulation of the neuronal electrical activity that may cause migraines. These findings identify TREK1 and TREK2 as potential molecular targets for migraine treatment and suggest that fsATI should be considered as a distinct class of mutations.
Mots clés : canaux ioniques / excitabilité / hétéromérisation / douleur / migraine
Key words: ion channels / excitability / heteromerization / pain / migraine
© Société de Biologie, 2019
Introduction
Il est difficile de comprendre pourquoi des mutations génétiques délétères du même gène se trouvent être ou ne pas être associées à une pathologie. La migraine est un désordre neurologique handicapant qui affecte 15 % de la population. Une attaque migraineuse se traduit par des maux de tête, ayant un caractère pulsatile, d’une durée de 4 à 72 h, accompagnés d’une hypersensibilité à de nombreux stimuli tels que la lumière ou le son pouvant mener à des nausées voire des vomissements. Les migraines sont cliniquement divisées en deux sous-types : la migraine avec aura (MA), quand elle est précédée de perturbations neurologiques transitoires, telles que des auras visuelles, et la migraine sans aura (MO). La dépression corticale envahissante est sous-jacente aux auras, bien que la relation de causalité n’ait pas encore été élucidée. La migraine est une maladie complexe liée, entre autres, à l’hyperexcitabilité électrique des neurones sensoriels de la face appelés neurones trigéminaux. Cette activation et la sensibilisation qui en découle mènent à un relargage de peptides pro-inflammatoires (Messlinger et al., 2011), composants clé dans l’initiation et la transmission de la migraine (Figure 1). Cette excitabilité électrique est sous le contrôle des canaux ioniques.
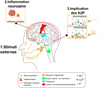 |
Figure 1 Représentation schématique soulignant les mécanismes qui contribuent à la pathogenèse de la migraine. 1. La migraine est déclenchée lorsqu’une vague électrique, initiée par des stimuli endogènes et exogènes, va perturber le système de modulation sensorielle dans le cerveau pour finalement activer les premières divisions du système trigéminal (V1/2/3). 2. Cette activation stimule le relargage de neuropeptides comme le CGRP, à l’origine d’une dilatation vasculaire et d’une inflammation. Il en résulte une facilitation de la transmission du signal douloureux par ces mêmes divisions jusqu’aux corps cellulaires situés dans les ganglions trigéminaux. 3. À ce niveau, des mutations dans le gène kcnk18 et les dérégulations conséquentes des canaux K2P potentialisent l’hyperexcitabilité neuronale et la transmission de la douleur. En haut : en condition physiologique, la protéine TRESK fonctionnelle contrôle activement la conductance potassique et stabilise le potentiel de membrane négatif, et par conséquent l’équilibre du sodium (Na+) et du calcium (Ca2+). En bas : la mutation de TRESK détruit cet équilibre en empêchant l’efflux de K+, pour finalement faciliter l’émission de potentiels d’actions, renforçant la transmission de l’information nociceptive et le relargage des différents neurotransmetteurs des voies correspondantes (adapté d’Albury et al., 2017). |
Les canaux ioniques
Les cellules constituant tout organisme sont délimitées par une bicouche lipidique appelée membrane plasmique. Celle-ci isole l’intérieur de la cellule du milieu extracellulaire. Les molécules chargées telles que les ions ne peuvent traverser cette barrière hydrophobe que par l’intermédiaire de protéines transmembranaires. Des protéines enzymatiques intégrées à cette membrane utilisent l’énergie de l’ATP pour générer une distribution asymétrique (gradient de concentration) des ions de part et d’autre de cette membrane. La pompe Na/K ATPase, qui est responsable de la mise en place des gradients de Na+ et K+, est l’exemple le plus connu. L’énergie électrochimique produite par ces gradients est utilisée par la cellule pour transporter des molécules (de l’extérieur vers l’intérieur et inversement), et pour générer des signaux électriques. Ces derniers nécessitent des structures membranaires particulières que l’on appelle canaux ioniques, lesquels sont le plus souvent spécifiques d’un seul type d’ion. Par exemple, lors de l’ouverture de canaux sélectifs du K+, ce cation, plus concentré à l’intérieur qu’à l’extérieur, a tendance à sortir, créant ainsi une différence de potentiel entre les milieux intra- et extra-cellulaires, qui peut être calculée par l’équation de Nernst. Dans une cellule non excitée, ou au repos, le potentiel de membrane est très proche du potentiel d’équilibre de l’ion K+ (Ek). La raison en est que, dans cet état, les canaux K+ sont pratiquement les seuls ouverts. Par conséquent, ce sont eux qui imposent le potentiel de repos. C’est la raison pour laquelle l’intérieur de la cellule est polarisé négativement au repos. À l’inverse, l’ouverture des canaux sélectifs pour le Na+ ou le Ca2+ entraîne une entrée de ces cations dans la cellule, ce qui déplace le potentiel de membrane vers des valeurs plus positives, traduisant une dépolarisation de la cellule. Les signaux électriques générés par ces gradients sont l’un des moyens mis en place par la cellule pour réguler son activité en réponse à différents types de stimuli. La première fonction physiologique parfaitement décrite pour les canaux ioniques est la génération et la propagation des signaux électriques (potentiel d’action, PA) le long des axones. Dès le début des années cinquante, Hodgkin et Huxley expliquèrent les mécanismes de la genèse et de la propagation du PA. En particulier, un PA n’est possible que grâce à l’association de plusieurs conductances ioniques : sodiques, potassiques et parfois calciques. À chacun de ces canaux est associée une phase du PA. La phase de dépolarisation est due à l’ouverture des canaux sodiques. Cette dépolarisation ouvre les canaux calciques dépendants du potentiel et responsables de la phase plateau. La phase de repolarisation est déclenchée par l’ouverture des canaux potassiques dépendants du potentiel de membrane et/ou du calcium.
Les canaux potassiques
Les canaux potassiques ne sont pas seulement impliqués dans la genèse d’un potentiel d’action, ils sont également à l’origine de nombreuses fonctions physiologiques. Les rôles physiologiques des canaux potassiques ont pu être mis en évidence grâce aux énormes progrès de la biologie moléculaire qui sont intervenus au cours de ces trente dernières années. L’émergence des techniques de clonage a permis l’étude de la régulation et des propriétés électrophysiologiques et pharmacologiques de nombreux canaux ioniques. Les canaux potassiques sont régulés par une grande variété de stimuli chimiques (pH, osmolarité, oxygène, anesthésiques), physiques (activité électrique, pression, température) et biologiques (ATP, calcium, protéine G, acides gras, kinases, phosphatases). Dans les cellules excitables, l’activité de ces canaux joue un rôle important dans le contrôle de l’excitabilité et dans la génération et la fréquence des PA. Ils sont impliqués dans divers processus physiologiques tels que la mémoire, la neuroprotection, le contrôle respiratoire, la contraction musculaire, la sécrétion d’hormones et de neurotransmetteurs, la régulation du rythme cardiaque. Certains de ces canaux sont la cible des anesthésiques généraux ou volatils. Dans les cellules non excitables, les canaux potassiques sont impliqués dans le contrôle du volume cellulaire, la prolifération et l’apoptose. Afin d’assurer toutes ces fonctions, il existe une importante diversité moléculaire de canaux potassiques présentant des modes de régulation distincts. En effet, il existe chez l’homme au moins 77 gènes qui codent pour ces canaux. Certains de ces canaux sont régulés par une dépolarisation cellulaire, d’autres par une déformation mécanique de la membrane ou encore par une élévation de la concentration de calcium intracellulaire, etc. Au niveau structural, les canaux potassiques sont des complexes protéiques transmembranaires. Chaque sous-unité composante possède un ou deux domaines P, et c’est l’assemblage de quatre domaines P qui permet de former un pore perméable aux ions (Figure 2). Le domaine P correspond à une séquence d’acides aminés entre deux hélices transmembranaires qui traverse partiellement la membrane plasmique. Cette partie transmembranaire contient le filtre de sélectivité aux ions potassium. Chez les mammifères, les sous-unités composant ces canaux contiennent deux, quatre ou six/sept segments transmembranaires. Les membres des familles de canaux à deux ou six/sept segments transmembranaires se caractérisent par la présence d’une seule boucle P alors que les canaux à quatre segments transmembranaires contiennent deux domaines P. Comme il faut quatre boucles P pour former un canal fonctionnel, les canaux à un domaine P (K1P, à deux ou six/sept segments transmembranaires) se tétramérisent en homo- ou hétéro-tétramère alors que les canaux à deux domaines P (K2P, à quatre segments transmembranaires) se dimérisent.
 |
Figure 2 Topologie de canaux potassiques KP. Les canaux voltage-dépendants (en rouge) possèdent six domaines transmembranaires et un domaine P (pore). Les canaux rectifiants entrants (en bleu) ont deux domaines transmembranaires et un domaine P. Les canaux à deux domaines P (K2P, en vert) possèdent quatre domaines transmembranaires et donc deux domaines pore (P1 et P2). |
La famille des canaux K2P dans les neurones
Ces canaux sont à l’origine d’un courant dit de fond. Le rôle principal de ce courant est la mise en place et le maintien du potentiel de repos des cellules et ainsi le contrôle de l’excitabilité cellulaire. En effet, quand ces canaux sont ouverts, ils laissent passer des ions K+ du milieu intracellulaire vers le milieu extracellulaire, ce qui entraîne un déplacement de charges positives de l’intérieur vers l’extérieur de la cellule, générant une hyperpolarisation cellulaire (cellule peu excitable). À l’inverse, la fermeture de ces canaux entraîne une dépolarisation de la cellule (cellule fortement excitable). Il existe quinze gènes codant pour ces canaux K2P. Les quinze membres de la famille partagent la même structure : quatre segments transmembranaires, deux boucles P et des extrémités carboxy- et amino-terminales intracellulaires. On peut classer ces quinze canaux en six grandes sous-familles en fonction des homologies de séquences et des propriétés fonctionnelles (Figure 3). La première sous-famille se compose des canaux TWIK1 (Tandem of P domains in Weak Inwardly rectifying K+ channel), TWIK2 et KCNK7 ; la deuxième se compose des canaux TREK1 (TWIK RElated K+ channel), TREK2 et TRAAK (TWIK Related Arachidonic Acid stimulated K+ channel) ; la troisième comprend TASK3 (TWIK-related Acid Sensing K+ channel), TASK1 et TASK5 ; la quatrième, THIK1 (TWIK related Halothane Inhibited K+ channel) et THIK2 ; la cinquième, TALK1 (TWIK-related Alcaline-pH-Activated K+ channel), TALK2 et TASK2 ; et la sixième sous-famille ne possède qu’un seul membre, le canal TRESK (TWIK-Related Spinal cord K+ channel). Ces différentes sous-familles de canaux se caractérisent par des propriétés fonctionnelles qui leur sont propres.
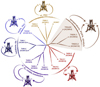 |
Figure 3 Dendrogramme de la famille des canaux potassiques à deux domaines P. L’effet de différents stimuli sur les courants de chaque sous-famille K2P est indiqué. PUFA, acides gras poly-insaturés ; PKC, protéine kinase C ; AA, acide arachidonique. |
Canaux potassiques à deux domaines P et neurones trigéminaux
Le système trigéminovasculaire est constitué de neurones trijumeaux et des vaisseaux (généralement cérébraux) qu’ils innervent. Les corps cellulaires bipolaires de ces neurones sont situés dans le ganglion du trijumeau (TG). Les axones périphériques projettent sur les structures crâniennes et les vaisseaux sanguins cérébraux alors que les axones projetant au niveau central atteignent notamment le noyau spinalis caudalis trigeminalis. L’activation des terminaisons sensorielles des neurones innervant les vaisseaux méningés provoque la libération de neuropeptides tels que la substance P et le CGRP. Il en résulte une vasodilatation et une inflammation qui sensibilisent les neurones trigéminaux impliqués dans la douleur. Cette sensibilisation permet à ces neurones de répondre anormalement à des stimulations qui sont normalement non douloureuses. Le message douloureux est alors transmis au niveau supérieur ce qui induit la migraine. L’hyperexcitabilité électrique de ces neurones sensoriels de la face joue donc un rôle clé dans la physiopathologie migraineuse.
L’excitabilité des neurones trigéminaux est notamment placée sous le contrôle de six canaux potassiques K2P : TASK1, TASK3, TREK1, TREK2, TRAAK et TRESK. L’importance de ces canaux dans le contrôle de l’excitabilité des neurones trigéminaux, et, ainsi, dans l’induction de la migraine, a récemment été mise en évidence par la découverte d’une version mutée du canal TRESK qui est liée à la migraine (Lafrenière et al., 2010).
Le canal TRESK et la migraine – « la controverse »
La protéine mutée TRESK-MT, résultant d’une délétion de deux paires de bases (F139WfsX24) dans le gène KCNK18, est en corrélation parfaite avec un phénotype migraineux sur plusieurs générations (Lafrenière et al., 2010). Cette mutation déplace le cadre de lecture et induit ainsi la formation d’un canal tronqué au niveau de sa partie carboxy-terminale. Ce canal mutant, en plus d’être non fonctionnel, agit comme dominant négatif du canal TRESK sauvage, de telle sorte que TRESK-MT abolit le courant généré par ce dernier. En accord avec l’implication de cette mutation dans la migraine, les neurones trigéminaux exprimant ce canal mutant voient leur excitabilité augmentée, ce qui explique l’induction de la migraine (Liu et al., 2013). Cependant, des études subséquentes ont décrit une autre mutation TRESK-C110R, qui, comme TRESK-MT, conduit à la formation d’un canal non fonctionnel agissant comme dominant négatif sur le canal sauvage (Andres-Enguix et al., 2012). Bien que TRESK-C110R et TRESK-MT semblent être similaires fonctionnellement, seule la mutation TRESK-MT est associée à une augmentation de l’excitabilité neuronale et à un phénotype migraineux. En effet, l’expression du mutant TRESK-C110R dans des neurones de ganglions trigéminaux n’induit pas une augmentation de l’excitabilité, ce qui pourrait expliquer pourquoi ce mutant est retrouvé à la fois chez des personnes migraineuses et chez les sujets contrôles (Guo et al., 2014). Comment deux mutations génétiques délétères du même gène TRESK peuvent-elles être, ou ne pas être, associées à la migraine ?
Le canal TRESK peut s’hétéromériser
Comme décrit précédemment, les canaux K2P sont actifs à l’état de dimère. La famille K2P est composée de 15 membres et certains d’entre eux sont capables de former des hétéro-dimères (Levitz et al., 2016). Nous avons émis l’hypothèse selon laquelle la différence entre les deux mutants TRESK-MT et TRESK-C110R pouvait provenir de leur capacité à réguler de manière différente les autres canaux K2P au travers de l’hétéromérisation.
L’utilisation de la technique de précipitation en molécule unique, le SiMPull (Jain et al., 2012), nous a permis de montrer que TRESK était capable de s’hétéromériser avec d’autres canaux K2P, notamment TREK1 et TREK2, aussi exprimés dans les neurones trigéminaux (Figure 4).
Ayant démontré que TRESK pouvait s’hétéromériser avec TREK1 et TREK2, nous avons testé la capacité des mutants TRESK (TRESK-MT et TRESK-C110R) à réguler TREK1 et TREK2. Les études fonctionnelles utilisant l’électrophysiologie révèlent que TRESK-MT, mais pas TRESK-C110R, est capable d’inhiber TREK1 et TREK2. Ainsi, le mutant impliqué dans la migraine, en plus de sa capacité à inactiver TRESK, peut inhiber TREK1 et TREK2, alors que le mutant TRESK-C110R, qui n’est pas impliqué dans la migraine, ne modifie pas les courants TREK1/2. Ce résultat nous a amenés à tester l’importance de TREK1 et TREK2 dans le contrôle de l’excitabilité des neurones trigéminaux et leur implication dans l’augmentation d’excitabilité induite par l’expression de TRESK-MT. Pour ce faire, nous avons généré des souris dont les gènes TREK1 et TREK2 sont invalidés. L’enregistrement de neurones trigéminaux des souris TREK1−/−/TREK2−/− a permis de montrer que l’absence d’expression de ces canaux génère un état basal hyperexcitable identique à celui observé dans les neurones exprimant TREK-MT. De plus, la surexpression de TRESK-MT dans ces neurones n’est plus capable d’augmenter leur excitabilité, démontrant que TRESK-MT augmente l’excitabilité via l’inhibition de TREK1 et TREK2. Ainsi, la différence entre le mutant impliqué dans la migraine et celui qui ne l’est pas réside dans la capacité du premier à inhiber TREK1 et TREK2. Les canaux TREK1 et TREK2 ayant un rôle de frein de l’excitabilité, il en résulte une augmentation de l’excitabilité des neurones trigéminaux induisant ainsi la migraine. Enfin, nous avons testé le comportement des souris dont TREK1 et TREK2 ont été invalidés génétiquement et avons trouvé qu’elles présentent un phénotype migraineux qui disparaît sous l’action de médicaments utilisés pour traiter les migraines chroniques. C’est donc l’inhibition de TREK1 et TREK2, mais pas de TRESK, qui conduit à la migraine, ouvrant ainsi de nouvelles pistes pour le traitement de cette pathologie.
 |
Figure 4 TRESK hétéromérise avec TREK1 et TREK2. Des cellules HEK exprimant GFP-TRESK et TREK1/2 fusionné à un marqueur HA sont lysées et immobilisées sur une lamelle passivée avec du polyéthylène glycol (PEG) conjugué avec un anticorps biotinylé anti-HA. La figure illustre les différentes étapes jusqu’à la visualisation des complexes TRESK-TREK (cf. Royal et al., 2019 pour la description complète des techniques utilisées). |
L’initiation alternative de la traduction induite par un déplacement du cadre de lecture
La mutation TRESK-MT est censée générer un canal tronqué non fonctionnel, appelé TRESK-MT1. Alors que le canal TRESK sauvage est capable d’interagir avec TREK1 et TREK2 dans un essai de précipitation en molécules uniques, TRESK-MT, dépourvu de la partie carboxy-terminale de TRESK, ne l’est pas. Si l’inhibition de TREK1/2 observée dans les cellules co-exprimant TRESK-MT ne vient pas de TRESK-MT1, il faut envisager l’implication d’un autre mécanisme. Normalement, un ARNm va permettre de produire une protéine. Mais dans certains cas, cette loi n’est pas suivie (Kozak, 1999). En effet, un site d’initiation de la traduction alternatif peut parfois être reconnu par les ribosomes et induire la synthèse d’une deuxième protéine à partir du même ARNm. Nous avons trouvé la présence d’un tel site d’initiation alternatif spécifiquement dans la séquence de TRESK-MT. Ce site alternatif a été mis en évidence par la délétion de deux paires de bases. Cette nouvelle protéine, appelée TRESK-MT2, correspond à un canal TRESK tronqué au niveau de sa partie N-terminale et donc non fonctionnel. L’existence de ce second transcrit a été mise en évidence par une série d’expériences de biologie cellulaire et de biochimie (Royal et al., 2019). Nous avons appelé fsATI (pour frameshift mutation-induced Alternative Translation Initiation) ce nouveau mécanisme de mise en place d’un site alternatif de la traduction par un déplacement du cadre de lecture (Figure 5). Nous avons testé, par une technique de précipitation en molécules uniques, la capacité de TRESK-MT2 à interagir avec TREK1 et TREK2 et avons trouvé que ce mutant s’associe fortement à ces deux canaux K2P. Nous avons alors recherché l’effet fonctionnel de cette interaction et avons trouvé que TRESK-MT2 était capable d’inhiber les canaux TREK1 et TREK2. En outre, les résultats obtenus sur les neurones trigéminaux sauvages montrent que l’inhibition de TREK1 et TREK2 par TRESK-MT2 induit une forte augmentation de l’excitabilité de ces neurones, similaire à celle observée dans les neurones dont TREK1 et TREK2 ont été invalidés.
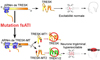 |
Figure 5 TRESK-MT provoque une migraine via l’inhibition de TREK1 et TREK2 par la protéine tronquée TRESK-MT2. En haut, excitabilité physiologique, normale, des neurones trigéminaux exprimant la forme sauvage de TRESK. En bas, les personnes portant la forme mutée du canal TRESK (TRESK-MT, symbolisée par une étoile noire) présentent une allodynie liée à la migraine. Le gène muté, par un mécanisme appelé fsATI, est traduit en deux protéines, TRESK-MT1 et TRESK-MT2. TRESK-MT1 va inhiber TRESK alors que TRESK-MT2 va cibler TREK1 et TREK2, menant à une hyperexcitabilité neuronale et à un phénotype migraineux. |
L’inhibition de TREK1 et TREK2 par TRESK-MT2 induit l’hyperexcitabilité
Ayant trouvé que l’inhibition de TREK1 et TREK2 était induite par TRESK-MT2, nous avons voulu savoir si l’expression de TRESK-MT2 était suffisante pour induire un phénotype migraineux, comme celui observé chez les patients portant la mutation TRESK-MT. Pour ce faire, nous avons surexprimé TRESK-MT2 dans les neurones trigéminaux grâce à des vecteurs viraux et avons testé le phénotype migraineux. La migraine peut être générée via l’utilisation de molécules donneuses d’oxyde nitrique comme l’isosorbide dinitrate. La migraine ainsi induite s’accompagne d’une hypersensibilité faciale, directement reliée à la sensibilité des nerfs trijumeaux innervant la face, qui peut être mesurée avec des filaments de von Frey. Ainsi, la sensibilité faciale de ces rats est utilisée comme un index reflétant le phénotype migraineux. Après une semaine d’infection, pour laisser le temps au virus d’exprimer TRESK-MT2, nous avons observé que les rats infectés présentaient une hypersensibilité faciale. Cette dernière est similaire à celle observée chez les rats soumis à une induction de migraine par les donneurs d’oxyde nitrique. Cette expérience confirme que la protéine TRESK-MT2 est suffisante pour induire le phénotype migraineux observé chez les patients.
TRESK-MT est-il le seul mutant de type fsATI ?
TRESK-MT est la première illustration d’une mutation qui induit la formation de deux protéines à partir d’un même ARN, au lieu d’une seule comme attendu, à la suite d’un déplacement du cadre de lecture. Afin de savoir si ce nouveau mécanisme de transmission de maladie héréditaire était, ou non, un cas isolé, nous avons cherché dans les bases de données publiques si d’autres mutations de type délétion de paires de bases pouvaient, comme la mutation TRESK-MT, créer un site alternatif de traduction. Nous avons trouvé une autre mutation du canal TRESK capable de créer un site alternatif d’initiation de la traduction. Nos données de biologie cellulaire et de biochimie confirment que cette mutation Y121LfsX44 conduit à la production de deux protéines (MT1 et MT2) et induit l’inhibition des canaux TREK1 et TREK2. Afin de savoir si cette mutation était liée à la maladie, nous avons alors confronté cette séquence aux bases de données cliniques et avons trouvé que cette mutation est la seule mutation de TRESK qui a également été liée à un phénotype migraineux dans des cohortes de populations asiatiques. La co-expression avec TREK1 dans un système d’expression hétérologue conduit à des résultats similaires : la production de deux protéines et l’inhibition des canaux TREK1 démontrant que la mutation TRESK-MT n’est pas un cas isolé.
Conclusion
Ces travaux démontrent que la différence entre les mutants du canal TRESK non fonctionnels qui sont – ou non – impliqués dans la migraine réside dans leur capacité à inhiber TREK1 et TREK2 en plus de TRESK. TREK1 et TREK2 constituent ainsi de nouvelles cibles pour le traitement de cette pathologie. D’autre part, ces travaux nous ont permis de découvrir un nouveau mécanisme de transmission de maladie génétique appelé « fsATI », qui doit maintenant être pris en compte dans l’étude des mutations impliquées dans les pathologies humaines.
Références
- Albury, C.L., Stuart, S., Haupt, L.M., Griffiths, L.R. (2017). Ion channelopathies and migraine pathogenesis. Mol Genet Genomics, 292, 729-739. [CrossRef] [PubMed] [Google Scholar]
- Andres-Enguix, I., Shang, L., Stansfeld, P.J., Morahan, J.M., Sansom, M.S.P., Lafrenière, R.G., Roy, B., Griffiths, L.R., Rouleau, G.A., Ebers, G.C., Cader Z.M., Tucker S.J. (2012). Functional analysis of missense variants in the TRESK (KCNK18) K channel. Sci Rep, 2, 237. [CrossRef] [PubMed] [Google Scholar]
- Guo, Z., Liu, P., Ren, F., Cao, Y.-Q. (2014). Nonmigraine-associated TRESK K+ channel variant C110R does not increase the excitability of trigeminal ganglion neurons. J Neurophysiol, 112, 568-579. [CrossRef] [PubMed] [Google Scholar]
- Jain, A., Liu, R., Xiang, Y.K., Ha, T. (2012). Single-molecule pull-down for studying protein interactions. Nat Protoc, 7, 445-452. [CrossRef] [PubMed] [Google Scholar]
- Kozak, M. (1999). Initiation of translation in prokaryotes and eukaryotes. Gene, 234, 187-208. [Google Scholar]
- Lafrenière, R.G., Cader, M.Z., Poulin, J.-F., Andres-Enguix, I., Simoneau, M., Gupta, N., Boisvert, K., Lafrenière, F., McLaughlan, S., Dubé, M.-P., Marcinkiewicz M.M., Ramagopalan S., Ansorge O., Brais B., Sequeiros J., Pereira-Monteiro J.M., Griffiths L.R., Tucker S.J., Ebers G., Rouleau G.A. (2010). A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat Med, 16, 1157-1160. [CrossRef] [PubMed] [Google Scholar]
- Levitz, J., Royal, P., Comoglio, Y., Wdziekonski, B., Schaub, S., Clemens, D.M., Isacoff, E.Y., Sandoz, G. (2016). Heterodimerization within the TREK channel subfamily produces a diverse family of highly regulated potassium channels. Proc Natl Acad Sci USA, 113, 4194-4199. [CrossRef] [Google Scholar]
- Liu, P., Xiao, Z., Ren, F., Guo, Z., Chen, Z., Zhao, H., Cao, Y.-Q. (2013). Functional analysis of a migraine-associated TRESK K+ channel mutation. J Neurosci, 33, 12810-12824. [CrossRef] [PubMed] [Google Scholar]
- Messlinger, K., Fischer, M.J.M., Lennerz, J.K. (2011). Neuropeptide effects in the trigeminal system: Pathophysiology and clinical relevance in migraine. Keio J Med, 60, 82-89. [CrossRef] [PubMed] [Google Scholar]
- Royal, P., Andres-Bilbe, A., Prado, P.Á., Verkest, C., Wdziekonski, B., Schaub, S., Baron, A., Lesage, F., Gasull, X., Levitz, J., Sandoz G. (2019). Migraine-associated TRESK mutations increase neuronal excitability through alternative translation initiation and inhibition of TREK. Neuron, 101, 232-245.e6. [CrossRef] [PubMed] [Google Scholar]
Citation de l’article : Royal, P., Ávalos Prado, P., Wdziekonski, B., et Sandoz, G. (2019). Canaux potassiques à deux domaines P (K2P) et migraine. Biologie Aujourd’hui, 213, 51-57
Liste des figures
|
Figure 1 Représentation schématique soulignant les mécanismes qui contribuent à la pathogenèse de la migraine. 1. La migraine est déclenchée lorsqu’une vague électrique, initiée par des stimuli endogènes et exogènes, va perturber le système de modulation sensorielle dans le cerveau pour finalement activer les premières divisions du système trigéminal (V1/2/3). 2. Cette activation stimule le relargage de neuropeptides comme le CGRP, à l’origine d’une dilatation vasculaire et d’une inflammation. Il en résulte une facilitation de la transmission du signal douloureux par ces mêmes divisions jusqu’aux corps cellulaires situés dans les ganglions trigéminaux. 3. À ce niveau, des mutations dans le gène kcnk18 et les dérégulations conséquentes des canaux K2P potentialisent l’hyperexcitabilité neuronale et la transmission de la douleur. En haut : en condition physiologique, la protéine TRESK fonctionnelle contrôle activement la conductance potassique et stabilise le potentiel de membrane négatif, et par conséquent l’équilibre du sodium (Na+) et du calcium (Ca2+). En bas : la mutation de TRESK détruit cet équilibre en empêchant l’efflux de K+, pour finalement faciliter l’émission de potentiels d’actions, renforçant la transmission de l’information nociceptive et le relargage des différents neurotransmetteurs des voies correspondantes (adapté d’Albury et al., 2017). |
| Dans le texte | |
|
Figure 2 Topologie de canaux potassiques KP. Les canaux voltage-dépendants (en rouge) possèdent six domaines transmembranaires et un domaine P (pore). Les canaux rectifiants entrants (en bleu) ont deux domaines transmembranaires et un domaine P. Les canaux à deux domaines P (K2P, en vert) possèdent quatre domaines transmembranaires et donc deux domaines pore (P1 et P2). |
| Dans le texte | |
|
Figure 3 Dendrogramme de la famille des canaux potassiques à deux domaines P. L’effet de différents stimuli sur les courants de chaque sous-famille K2P est indiqué. PUFA, acides gras poly-insaturés ; PKC, protéine kinase C ; AA, acide arachidonique. |
| Dans le texte | |
|
Figure 4 TRESK hétéromérise avec TREK1 et TREK2. Des cellules HEK exprimant GFP-TRESK et TREK1/2 fusionné à un marqueur HA sont lysées et immobilisées sur une lamelle passivée avec du polyéthylène glycol (PEG) conjugué avec un anticorps biotinylé anti-HA. La figure illustre les différentes étapes jusqu’à la visualisation des complexes TRESK-TREK (cf. Royal et al., 2019 pour la description complète des techniques utilisées). |
| Dans le texte | |
|
Figure 5 TRESK-MT provoque une migraine via l’inhibition de TREK1 et TREK2 par la protéine tronquée TRESK-MT2. En haut, excitabilité physiologique, normale, des neurones trigéminaux exprimant la forme sauvage de TRESK. En bas, les personnes portant la forme mutée du canal TRESK (TRESK-MT, symbolisée par une étoile noire) présentent une allodynie liée à la migraine. Le gène muté, par un mécanisme appelé fsATI, est traduit en deux protéines, TRESK-MT1 et TRESK-MT2. TRESK-MT1 va inhiber TRESK alors que TRESK-MT2 va cibler TREK1 et TREK2, menant à une hyperexcitabilité neuronale et à un phénotype migraineux. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.