| Issue |
Biologie Aujourd’hui
Volume 215, Number 3-4, 2021
|
|
|---|---|---|
| Page(s) | 107 - 118 | |
| DOI | https://doi.org/10.1051/jbio/2021010 | |
| Published online | 11 mars 2022 | |
Article
Mécanismes d’action et rôles multiples des β-arrestines dans la biologie des récepteurs couplés aux protéines G
β-arrestins, their mechanisms of action and multiple roles in the biology of G protein-coupled receptors
1
CNRS, IFCE, INRAE, Université de Tours, PRC,
37380
Nouzilly, France
2
Inria, Centre de recherche Inria Saclay-Île-de-France,
91120
Palaiseau, France
* Auteur correspondant : Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Reçu :
7
Septembre
2021
Résumé
La stimulation des récepteurs couplés aux protéines G (RCPG) induit des réponses biologiques à un large éventail de signaux extracellulaires. Les protéines G hétérotrimériques, qui sont recrutées aux RCPG actifs, conduisent à la génération de divers seconds messagers diffusibles. En plus des protéines G, seules deux familles de protéines présentent également la caractéristique remarquable de reconnaître la conformation active de la majorité des RCPG et de s’y lier : les kinases spécifiques des RCPG (GRK) et les β-arrestines. Ces deux familles de protéines ont initialement été identifiées en tant qu’acteurs clefs de la désensibilisation de l’activation des protéines G par les RCPG. Au fil des années, les β-arrestines ont été impliquées dans un nombre croissant d’interactions avec des protéines non réceptrices, élargissant le panel des fonctions cellulaires dans lesquelles elles sont impliquées. Il est maintenant bien établi que les β-arrestines, en échafaudant et en recrutant des complexes protéiques de manière dépendante de l’agoniste, régulent directement le trafic et la signalisation des RCPG. Des avancées remarquables ont été réalisées au cours des dernières années qui ont permis i) d’identifier des ligands biaisés capables, en stabilisant des conformations particulières d’un nombre croissant de RCPG, d’activer ou de bloquer l’action des β-arrestines indépendamment de celle des protéines G, certains de ces ligands présentant un intérêt thérapeutique ; ii) de mettre en évidence le rôle des β-arrestines dans la compartimentalisation de la signalisation des RCPG au sein de la cellule, en particulier depuis les endosomes, et, iii) de comprendre les détails moléculaires de leur interaction avec les RCPG et de leur activation grâce à des approches structurales et biophysiques.
Abstract
The stimulation of G protein-coupled receptors (GPCRs) induces biological responses to a wide range of extracellular cues. The heterotrimeric G proteins, which are recruited to the active conformation of GPCRs, lead to the generation of various diffusible second messengers. Only two other families of proteins exhibit the remarkable characteristic of recognizing and binding to the active conformation of most GPCRs: GPCR kinases (GRKs) and β-arrestins. These two families of proteins were initially identified as key players in the desensitization of G protein activation by GPCRs. Over the years, β-arrestins have been implicated in an increasing number of interactions with non-receptor proteins, expanding the range of cellular functions in which they are involved. It is now well established that β-arrestins, by scaffolding and recruiting protein complexes in an agonist-dependent manner, directly regulate the trafficking and signaling of GPCRs. Remarkable advances have been made in recent years which have made it possible i) to identify biased ligands capable, by stabilizing particular conformations of a growing number of GPCRs, of activating or blocking the action of β-arrestins independently of that of G proteins, some of these ligands holding great therapeutic interest; ii) to demonstrate β-arrestins’ role in the compartmentalization of GPCR signaling within the cell, and iii) to understand the molecular details of their interaction with GPCRs and of their activation through structural and biophysical approaches.
Mots clés : β-arrestines / signalisation intracellulaire / trafic intracellulaire / biais pharmacologiques / RCPG
Key words: β-arrestins / intracellular signaling / intracellular trafficking / pharmacological biases / GPCR
© Société de Biologie, 2022
Introduction
Les cellules sont exposées à une combinaison complexe de stimuli qui varient selon leurs natures chimiques, leurs concentrations et leurs durées d’exposition. En conséquence, les cellules ont développé des systèmes sophistiqués permettant la réception et l’interprétation des stimuli extracellulaires. Au cœur de ces systèmes, les récepteurs couplés aux protéines G (RCPG) représentent la famille de récepteurs membranaires la plus vaste et la plus polyvalente. Lors de la stimulation par l’agoniste, les RCPG subissent des changements de conformation qui exposent des sites de liaison intracellulaires pour des protéines de transduction. Typiquement, plusieurs voies de signalisation intracellulaires peuvent être activées en parallèle. Pour que des réponses biologiques appropriées (e.g., prolifération, différenciation, migration ou apoptose) puissent être apportées en fonction des situations physiologiques rencontrées, des mécanismes d’orchestration efficaces sont nécessaires. Même si de nombreuses protéines interagissent directement avec les RCPG, seules trois familles de protéines ont la capacité d’interagir spécifiquement avec les RCPG en conformation active : les protéines G hétérotrimériques qui ont donné leur nom aux RCPG, les kinases spécifiques des RCPG (GRK) et les β-arrestines (Reiter & Lefkowitz, 2006 ; DeWire et al., 2007 ; Reiter et al., 2012).
Les arrestines constituent une famille de quatre membres numérotés de 1 à 4. L’expression de l’arrestine 1 et de l’arrestine 4 est limitée aux bâtonnets et aux cônes rétiniens, respectivement. En revanche, l’arrestine 2 et l’arrestine 3, mieux connues sous le nom de β-arrestine 1 et β-arrestine 2, sont exprimées de manière ubiquiste (DeWire et al., 2007). Il est largement admis que les GRK et les β-arrestines orchestrent les activités des RCPG à trois niveaux différents : i) l’arrêt du signal induit par le découplage fonctionnel entre le récepteur et les protéines G par un mécanisme connu sous le nom de « désensibilisation homologue », ii) le trafic intracellulaire qui se traduit par l’internalisation, le recyclage et/ou la dégradation des récepteurs et iii) la signalisation qui conduit à l’activation ou l’inhibition de voies de signalisation intracellulaires, non seulement depuis la membrane plasmique, mais aussi à partir de compartiments intracellulaires.
Implication des β-arrestines dans la désensibilisation, l’internalisation, le trafic intracellulaire et la signalisation des RCPG
Depuis leur découverte il y a plus de trente ans, la perception des fonctions des β-arrestines n’a cessé de s’élargir, au point qu’elles sont maintenant indissociablement liées à tous les aspects clés de la fonction des RCPG. L’activation, la désensibilisation et l’internalisation de la majorité des RCPG non rétiniens sont régulées de manière critique par les deux β-arrestines. Deux éléments déclencheurs principaux contrôlent le recrutement de la β-arrestine aux RCPG : la modification induite par l’agoniste de la conformation du récepteur et la phosphorylation, médiée par les GRK, du récepteur occupé par le ligand (Gurevich & Benovic, 1993 ; Reiter et al., 2012). La première étape de l’activation du récepteur, la liaison au ligand, est elle-même impactée par les β-arrestines. L’augmentation allostérique de l’affinité de liaison d’un ligand lorsque le récepteur est complexé avec sa protéine G a été conceptualisée il y a plus de 35 ans dans le « modèle de complexe ternaire » (De Lean et al., 1980) et a été, par la suite, étayée par des preuves structurelles directes (DeVree et al., 2016). De façon intéressante, il a été démontré plus récemment que le recrutement de la β-arrestine à un RCPG induit un effet allostérique positif très similaire sur la liaison du ligand, soutenant l’existence d’un complexe ternaire alternatif impliquant la β-arrestine (Martini et al., 2002 ; Strachan et al., 2014).
Dès leur découverte, les β-arrestines ont été associées à l’atténuation du couplage des protéines G (DeWire et al., 2007). En effet, le RCPG actif est phosphorylé dans son extrémité C-terminale par les GRK et recrute alors la β-arrestine avec une affinité élevée. Cette interaction conduit à l’inhibition du couplage des protéines G par encombrement stérique (Reiter & Lefkowitz, 2006). Ce processus, généralement appelé « désensibilisation homologue », s’applique à la plupart des RCPG (Freedman & Lefkowitz, 1996). Il a été démontré plus tard que les β-arrestines ont également la capacité de recruter des enzymes de dégradation des seconds messagers, telles que les AMPc phosphodiestérases ou les diacylglycérol kinases, au récepteur actif (Perry et al., 2002 ; Nelson et al., 2007). Cette propriété remarquable implique que les β-arrestines désensibilisent doublement les RCPG : i) en inhibant le couplage des protéines G et ii) en augmentant simultanément le taux de dégradation du second messager à proximité du récepteur actif.
En plus de leur rôle dans la désensibilisation, les β-arrestines jouent également un rôle central dans l’internalisation des RCPG en interagissant avec des éléments clés de la machinerie endocytaire tels que la clathrine (Goodman et al., 1996), l’adaptateur de clathrine AP2 (Laporte et al., 1999), la petite protéine G ARF6 et son facteur d’échange de nucléotide guanine, ARNO (Claing et al., 2001), ainsi que la protéine de fusion sensible au N-éthylmaléimide (NSF) (McDonald et al., 1999). De plus, MDM2, une ubiquitine ligase E3, se lie aux β-arrestines et catalyse leur ubiquitination, ce qui est essentiel pour l’endocytose du RCPG médiée par la clathrine (Shenoy et al., 2001). La présence ou l’absence de clusters de sérine et de thréonine dans l’extrémité C-terminale du RCPG conditionne l’affinité du recrutement de la β-arrestine et affecte ainsi le trafic intracellulaire d’un grand nombre de RCPG (Oakley et al., 2000, 2001). De plus, bien que le mécanisme de recrutement des β-arrestines 1 et 2 aux RCPG soit similaire, l’affinité des RCPG pour la β-arrestine 1 ou 2 n’est pas obligatoirement équivalente. En effet, il a été défini deux classes de RCPG en fonction de leur affinité pour les deux isoformes de β-arrestine (Oakley et al., 2000). Les RCPG de classe A (récepteur β2 adrénergique, µ opioïde, …) ont une affinité plus grande pour la β-arrestine 2 que pour la β-arrestine 1 ainsi qu’une affinité nulle pour les arrestines visuelles. En revanche, les récepteurs de classe B (récepteur de l’angiotensine II, récepteur de la vasopressine, …) ont une affinité similaire pour les deux isoformes de β-arrestine ainsi qu’une affinité plus faible mais néanmoins existante pour les arrestines visuelles.
Au-delà de leurs rôles dans le contrôle de la désensibilisation et de l’internalisation, les β-arrestines sont désormais considérées comme des protéines de transduction (Lefkowitz & Shenoy, 2005 ; Reiter & Lefkowitz, 2006). Il a été largement documenté que les β-arrestines sont des échafaudages multifonctionnels qui interagissent avec de nombreux partenaires protéiques, y compris les protéines kinases, et qu’elles ont ainsi un impact sur la phosphorylation de nombreuses cibles intracellulaires (Xiao et al., 2007, 2010). Au fil des ans, plusieurs approches ont été utilisées pour déchiffrer les contributions respectives des protéines G et des β-arrestines à la fonction des RCPG. Il s’agit notamment i) d’invalidations des GRK ou des β-arrestines chez la souris et de l’utilisation de leurs homologues cellulaires MEF dérivés, ii) du blocage sélectif de la protéine G et des constituants de la voie de la β-arrestine via des ARN interférents, iii) de l’utilisation de dominants négatifs ou encore de petites molécules inhibitrices. Ces outils ont été utilisés avec succès pour décrypter de nouveaux mécanismes de transduction du signal et mieux caractériser la pharmacologie de RCPG spécifiques (DeWire et al., 2007 ; DeWire & Violin, 2011). L’émergence récente de la technologie CRISPR Cas9 a cependant conduit deux groupes à contester la capacité des β-arrestines à induire la signalisation indépendamment des protéines G, suggérant que les β-arrestines ne seraient que de simples rhéostats de l’action des protéines G (O’Hayre et al., 2017 ; Grundmann et al., 2018). Mais il a ensuite été démontré que les lignées CRISPR Cas9 conduisent à un recâblage des voies de signalisation par rapport aux lignées parentales d’origine (Luttrell et al., 2018). L’importance relative des β-arrestines dans la désensibilisation et la signalisation varie donc fortement en fonction du contexte cellulaire, du récepteur et du ligand. En conséquence, une analyse exhaustive et précise des différentes composantes de l’action des β-arrestines et des compensations qui font suite à l’édition du génome est indispensable pour pouvoir interpréter correctement les résultats de signalisation obtenus dans ce type de modèles cellulaires.
Le mécanisme de signalisation dépendant des β-arrestines le mieux caractérisé à ce jour est certainement celui des MAPK ERK1/2. Il a été démontré que les β-arrestines échafaudent Raf-1, MEK1 et ERK et séquestrent ERK1/2 phosphorylées dans le cytosol (Luttrell et al., 2001). De façon intéressante, les kinases ERK1/2 sont simultanément activées par la protéine G et par les β-arrestines par des mécanismes distincts. L’activation de ERK1/2 dépendante de la protéine G est rapide et généralement transitoire. En revanche, l’activation de ERK1/2 dépendante de la β-arrestine est plus lente mais prolongée. Cependant, dans certains cas, l’activation de ERK1/2 par la protéine G peut également inclure une phase soutenue, de sorte que la cinétique seule ne peut pas toujours distinguer la signalisation ERK1/2 médiée par la protéine G de celle impliquant la β-arrestine (Luo et al., 2008). En plus des MAPK ERK, de nombreuses autres voies de signalisation sont affectées par les β-arrestines (Ahn et al., 2020). En effet, les β-arrestines favorisent l’assemblage et l’activation des modules MAPK ASK1, MKK4/7 et JNK3 (McDonald et al., 2000) ainsi que MKK4, MKK7 et JNK1/2 (Kook et al., 2013) et déclenchent la signalisation p38 (Sun et al., 2002 ; Bruchas et al., 2006). La transactivation du récepteur EGF par les RCPG peut être régulée par les β-arrestines via l’activation d’une métalloprotéase matricielle transmembranaire qui clive le ligand EGF lié à la membrane (Noma et al., 2007). Les β-arrestines peuvent inhiber la signalisation NF-κB par stabilisation de IκBα (Gao et al., 2004). La β-arrestine 1 peut impacter directement les modifications épigénétiques en interagissant au niveau nucléaire avec les histones acétylases et désacétylases qui régulent la structure de la chromatine (Kang et al., 2005). D’autres mécanismes de signalisation médiés par la β-arrestine incluent, entre autres, la formation de fibres de stress dépendantes de RhoA (Barnes et al., 2005), la déphosphorylation d’Akt par la protéine phosphatase 2A (Beaulieu et al., 2005), l’induction de la traduction protéique dépendante de MNK (DeWire et al., 2008) et les effets anti-apoptotiques dépendant de p90RSK (Ahn et al., 2009), l’activation de la phospholipase A2 par la phosphatidylinositol 3-kinase (Walters et al., 2009) et l’activation de PTEN en aval de RhoA/ROCK (Lima-Fernandes et al., 2011).
Au niveau moléculaire, il a été montré, en utilisant différentes approches expérimentales, que les β-arrestines 1 et 2 subissent des changements de conformation lors de l’interaction avec l’extrémité carboxyle phosphorylée des récepteurs (Xiao et al., 2004 ; Charest et al., 2005 ; Nobles et al., 2007). Il a été proposé que les conformations de récepteurs fonctionnellement spécifiques induites par un ligand puissent être traduites en conformations spécifiques de la β-arrestine et avoir un impact sur leurs activités intracellulaires (Shukla et al., 2008). Ce point de vue a été exploré plus avant à l’aide de sondes intracellulaires BRET ou FRET capables de détecter le répertoire conformationnel de la β-arrestine avec une meilleure précision (Lee et al., 2016 ; Nuber et al., 2016). Ces études ont conclu que des conformations distinctes de la β-arrestine peuvent être stabilisées d’une manière spécifique au récepteur et/ou au ligand. De façon intéressante, il a été rapporté que différents sous-types de GRK exercent des fonctions de régulation spécialisées. Il a ainsi été démontré pour plusieurs RCPG que la génération de seconds messagers est atténuée par GRK2 mais non affectée par GRK5 ou GRK6, alors que l’activation de ERK dépendante de la β-arrestine 2 nécessite l’action de GRK5 et GRK6 (Iwata et al., 2005 ; Kim et al., 2005 ; Ren et al., 2005 ; Kara et al., 2006 ; Shenoy et al., 2006 ; Zidar et al., 2009). À la lumière de ces résultats, l’hypothèse selon laquelle il existe un « code-barres » de phosphorylation a été émise par les GRK à l’extrémité C terminale des RCPG. Ce code de phosphorylation régulerait la nature des fonctions intracellulaires des β-arrestines (Kim et al., 2005 ; Reiter & Lefkowitz, 2006 ; Shenoy et al., 2006 ; Tobin et al., 2008). Des études indépendantes ont démontré que la phosphorylation des RCPG est en effet préférentiellement dirigée vers des sites spécifiques d’une manière dépendante du ligand et de la kinase (Busillo et al., 2010 ; Butcher et al., 2011 ; Nobles et al., 2011 ; Yan et al., 2011 ; Heitzler et al., 2012).
Modifications post-transcriptionnelles des β-arrestines
Les arrestines, tout comme les RCPG, subissent diverses modifications post-transcriptionnelles qui influencent leur interaction avec les RCPG et avec d’autres protéines accessoires et affectent en particulier le trafic intracellulaire des récepteurs.
Phosphorylation
Deux études publiées en 1997 (Lin et al., 1997) et 2002 (Lin et al., 2002) ont montré que les β-arrestines sont phosphorylées à l’état basal. La stimulation du récepteur β2 adrénergique favorise leur déphosphorylation, plus particulièrement celle de la sous-population de β-arrestines colocalisées avec le récepteur à la membrane plasmique (Lin et al., 1997). Les sites de phosphorylation et les kinases ne sont pas conservés entre β-arrestine 1 et β-arrestine 2. La β-arrestine 1 est phosphorylée sur la Ser412 par les kinases ERK1/2 (Lin et al., 1997, 1999) tandis que la β-arrestine 2 est phosphorylée sur la Thr383 par la caséine kinase de type II et sur la Ser361 par une kinase inconnue (Lin et al., 2002). En utilisant les mutants non phosphorylable β-arrestine-1-S412A et phosphomimétique S412D, les auteurs ont montré que le recrutement d’arrestine au récepteur ainsi que la désensibilisation de ce dernier ne sont pas dépendants du statut de phosphorylation de l’arrestine. En revanche, le mutant S412A favorise la formation du complexe arrestine/clathrine ainsi que l’internalisation du récepteur (Lin et al., 1997). Des résultats similaires ont été obtenus en mutant les sites de phosphorylation de la β-arrestine 2 (Lin et al., 2002). Toutefois, une étude ultérieure, bien qu’ayant confirmé la phosphorylation de la β-arrestine 2 sur la Thr383 par CKII, n’a pas confirmé l’impact de cette phosphorylation sur la formation du complexe arrestine/clathrine (Kim et al., 2002). Plus récemment, un mécanisme moléculaire original impliquant la phosphorylation de la β-arrestine 2 sur la Thr383 par MEK en réponse à l’agoniste a été mis en évidence. Cette phosphorylation induirait un changement de conformation de la β-arrestine 2 qui apparaît comme un préalable à l’induction de la phosphorylation de ERK par plusieurs RCPG (Cassier et al., 2017). Une autre étude a rapporté un nouveau mécanisme de régulation de la dynamique d’interaction du complexe récepteur/β-arrestine 2 au niveau des endosomes. Cette modulation est médiée par la phosphorylation du résidu Thr178 de la β-arrestine 2 par ERK1/2 qui stabilise le complexe arrestine/récepteur et inhibe le recyclage de ce dernier à la membrane plasmique. Toutefois, le site Thr178 est présent uniquement dans la protéine β-arrestine 2 de rat et n’est pas conservé dans la forme humaine. Notons que les deux mutants phosphomimétiques β-arrestine-2-T178D (pour le rat) et -K178D (pour l’humain) altèrent de manière similaire le trafic des récepteurs (Khoury et al., 2014). Ces études montrent que la voie ERK1/2, une des voies de signalisation majeures en aval des RCPG, permet l’activation de boucles de rétroaction qui modulent le trafic et donc l’activité signalétique des RCPG.
Ubiquitination
C’est via leur ubiquitination que les arrestines contrôlent la vitesse de dégradation ou de recyclage des récepteurs. Ainsi, l’ubiquitination d’arrestine par l’E3-ligase Mdm2 favorise l’internalisation du récepteur β2 adrénergique (Shenoy et al., 2001). De manière intéressante, les sites ubiquitinés sur les β-arrestines varient selon le récepteur activé et pourraient donc être impliqués dans différentes fonctions physiologiques (Perroy et al., 2004 ; Shenoy et al., 2007). En revanche, le même schéma d’ubiquitination semble être partagé entre les récepteurs de classe A et les récepteurs de classe B. Les β-arrestines engagées aux récepteurs de classe A, capables de recycler à la membrane plasmique, sont ubiquitinées de manière transitoire, contrairement aux arrestines en complexe avec les récepteurs de classe B dirigés vers les voies de dégradation lysosomales, dont l’ubiquitination est plus soutenue (Perroy et al., 2004 ; Shenoy et al., 2007). Cette ubiquitination soutenue semble aussi favoriser la formation de signalosomes au niveau des arrestines, qui engagent par exemple des composants de la voie ERK1/2 (Shenoy et al., 2007).
SUMOylation
La SUMOylation de la β-arrestine 2 sur les résidus K295 et K400 a été démontrée (Wyatt et al., 2011). La SUMOylation du site majeur K400 est activée par la stimulation du récepteur β2 adrénergique. Les auteurs ont émis l’hypothèse selon laquelle le changement de conformation de la β-arrestine lors de sa fixation au récepteur activé permet d’exposer le résidu K400 de la queue C-ter à l’enzyme Ubc9. L’inhibition de la SUMOylation de la β-arrestine, par mutagénèse dirigée ou par baisse de l’expression d’Ubc9, bloque l’internalisation des récepteurs AT1R de l’angiotensine et du récepteur β2 adrénergique. Cette inhibition ne correspond pas à une baisse du recrutement de la β-arrestine au récepteur mais à une diminution de son interaction avec AP-2, un des composants de la machinerie endocytique.
S-nitrosylation
L’oxyde nitrique (NO) participe à la S-nitrosylation des résidus Cys des protéines cibles en convertissant le groupement thiol en S-nitrosothiol (S-NO) (Gould et al., 2013). Il a été démontré que la β-arrestine 2, mais pas la β-arrestine 1, est S-nitrosylée sur le résidu Cys410 après stimulation du récepteur β2 adrénergique (Ozawa et al., 2008). Cette S-nitrosylation entraîne le découplage du complexe eNOS/arrestine et favorise la liaison d’arrestine à AP-2 et la clathrine, ce qui induit l’internalisation du récepteur β2 adrénergique. Après stimulation, la β-arrestine 2 est rapidement dénitrosylée. Il est intéressant de noter que dans ce processus, la β-arrestine 2 sert aussi de protéine d’échafaudage pour l’enzyme eNOS activée par le récepteur β2 adrénergique (Ozawa et al., 2008) et facilite la S-nitrosylation d’autres substrats tels que GRK2 (Whalen et al., 2007). Ainsi, eNOS pourrait permettre la coordination de la régulation de trois éléments majeurs dans l’internalisation et le trafic des RCPG : les GRK, les arrestines et la dynamine.
β-arrestines et signalisation biaisée
De nombreuses évidences structurelles, biophysiques et pharmacologiques accumulées depuis quelques années ont profondément transformé notre vision de l’activation des RCPG et de leur ciblage thérapeutique. Il n’y a pas si longtemps, un large consensus existait pour penser qu’une conformation inactive d’un récepteur est en équilibre avec une unique conformation active liée à un ligand. En conséquence, la force d’un agoniste était censée refléter directement la proportion de conformation de récepteur active par rapport à la conformation inactive. Par la suite, la découverte d’agonistes partiels et d’agonistes inverses a révélé de nouvelles propriétés pharmacologiques au-delà des agonistes complets et des antagonistes neutres, mais ces types d’activités étaient toujours compatibles avec le modèle à deux états. Cependant, plusieurs exemples ont été rapportés qui ne correspondaient pas à ce paradigme : certains composés ont généré différentes puissances relatives dans différents dosages (Watson et al., 2000). Ces résultats, controversés au départ, ont été répétés avec un nombre croissant de RCPG. Entre-temps, le fait que plusieurs conformations de récepteurs actives et inactives coexistent a été étayé par des preuves structurelles et biophysiques très fortes (Kobilka, 2011 ; Nygaard et al., 2013 ; Wacker et al., 2013). Par conséquent, la théorie pharmacologique a été révisée et l’efficacité est désormais considérée comme multidimensionnelle. Elle intègre explicitement la notion selon laquelle les récepteurs engagent des sous-ensembles distincts de leur répertoire de signalisation complet (Galandrin et al., 2007). Cela signifie que différents sous-ensembles de conformations peuvent être stabilisés par différents agonistes ou mutation/polymorphisme pour un RCPG donné et que chacun de ces ensembles conformationnels est connecté à des mécanismes de transduction distincts. C’est le concept de signalisation sélective ou biais pharmacologique (Kenakin, 2003 ; Violin & Lefkowitz, 2007 ; Reiter et al., 2012). Selon ces principes, il est possible de contrôler sélectivement l’activation de telle ou telle voie de signalisation avec des ligands biaisés ou avec des modifications spécifiques d’acides aminés au niveau des récepteurs. Les sites orthostériques sur les RCPG se lient aux agonistes endogènes et sont également reconnus par les antagonistes compétitifs classiques et les agonistes inverses. En revanche, les sites allostériques sur un récepteur sont distincts du site orthostérique et peuvent affecter soit positivement soit négativement l’activité du récepteur, conjointement avec des ligands orthostériques ou seuls. Il est important de noter que, pour les RCPG, les modulateurs allostériques synthétiques sont maintenant découverts à un rythme élevé et peuvent également entraîner un biais pharmacologique, offrant de nouvelles voies dans la découverte de médicaments (Changeux & Christopoulos, 2016). Ces ligands allostériques peuvent moduler les conformations des récepteurs en présence de ligands orthostériques et ont donc le potentiel d’affiner, positivement ou négativement, les réponses suscitées aussi bien par des ligands endogènes ou synthétiques.
L’étude des biais pharmacologiques est rapidement devenue un domaine de recherche extrêmement actif et, une fois de plus, les β-arrestines y occupent une place prépondérante puisqu’un grand nombre de ligands présentant des biais sur les fonctions médiées par la β-arrestine ont été rapportés. Certains ligands biaisés favorisent la transduction dépendante de la protéine G tandis que d’autres déclenchent préférentiellement des voies médiées par la β-arrestine. Il est important de noter que des ligands biaisés capables de stabiliser un sous-ensemble du répertoire de conformations du récepteur ont été rapportés pour améliorer l’équilibre entre les effets secondaires indésirables et les effets bénéfiques (Whalen et al., 2011). L’avènement de nouvelles classes non conventionnelles de composés ciblant les RCPG tels que les pepducines (Carr & Benovic, 2016), les aptamères (Kahsai et al., 2016), les intracorps (Staus et al., 2014) ou les nanobodies (Mujic-Delic et al., 2014 ; Staus et al., 2016) étendent encore plus l’éventail des possibilités d’approches innovantes de découverte de médicaments à développer à l’avenir.
Jusqu’à présent, les ligands biaisés ont concentré l’essentiel de l’attention dans le domaine de la pharmacologie RCPG car ils représentent des pistes potentielles pour le développement de nouveaux médicaments. Cependant, la notion de biais s’applique également aux modifications se produisant au niveau du récepteur (Landomiel et al., 2014). Les premiers exemples de mutations conduisant à un biais β-arrestine ont concerné le récepteur de l’angiotensine de type 2 : DRY-AAY (Wei et al., 2003) et le récepteur β2 adrénergique : β2AR-TYY (Shenoy et al., 2006). Cette notion de biais pharmacologique joue donc également un rôle crucial en médecine car elle peut se matérialiser chez les patients portant des mutations ou des polymorphismes critiques au niveau des RCPG. De ce fait, le concept de biais pharmacologique change la façon d’étudier les conséquences fonctionnelles des mutations et des polymorphismes se produisant au niveau du récepteur. Ce type de question était traditionnellement évalué en suivant la perte ou le gain de fonction selon le modèle simple à deux états. À présent, l’exploration doit intégrer les multiples dimensions de l’activité des récepteurs grâce à des analyses multiplexées des différentes voies de signalisation induites par l’activation des récepteurs en aval.
Rôle des β-arrestines dans la compartimentalisation de la signalisation des RCPG
Une énigme majeure associée à la signalisation RCPG réside dans le fait que le nombre de ligands et de récepteurs semble largement surpasser le nombre relativement limité de mécanismes de transduction et de voies de signalisation en aval disponibles. Pour contourner ce problème, il a été proposé que la machinerie de signalisation puisse utiliser un encodage spatial et temporel afin de maintenir l’ensemble des informations et la spécificité conférés par la paire récepteur/ligand (Lohse & Hofmann, 2015). Dans cette optique, les événements de signalisation déclenchés par un RCPG sont caractérisés par leur cinétique et leurs schémas spatiaux et correspondent à une signature spécifique du récepteur, du contexte cellulaire et de la nature du ligand. Le régime d’exposition au ligand peut également conduire à des signatures de signalisation spécifiques, une possibilité qui pourrait être particulièrement pertinente dans le contexte des systèmes endocriniens. Les aspects cinétiques et spatiaux de la signalisation RCPG ont commencé à être explorés avec l’élucidation de la dynamique de l’activation des récepteurs, du couplage des protéines G et de l’activation des protéines G pour certains récepteurs (Lohse et al., 2008 ; Jensen et al., 2009). Dans le modèle classique, la signalisation de la protéine G provient de la surface cellulaire et est suivie d’une extinction rapide, médiée par la β-arrestine, de la signalisation de la protéine G. Des découvertes récentes ont commencé à remettre en question ce paradigme. Il a été rapporté qu’un certain nombre de RCPG provoquent une signalisation soutenue de la protéine G, plutôt que d’être désensibilisés après une stimulation agoniste initiale (Calebiro et al., 2009 ; Ferrandon et al., 2009 ; Mullershausen et al., 2009 ; Feinstein et al., 2013 ; Irannejad et al., 2013). Par exemple, il a proposé que, pour certains RCPG, une série de vagues de signalisation distinctes puisse apparaître lors de l’activation (Lohse & Calebiro, 2013). Dans ce modèle, une première vague est déclenchée à la surface cellulaire lors du couplage de la protéine G et entraîne la libération du second messager. Une deuxième vague suit au niveau des puits recouverts de clathrine lorsque la β-arrestine associée au récepteur induit des signaux tels que l’activation d’ERK. Récemment, une troisième vague a été décrite qui implique la signalisation via les protéines G du compartiment endosomal et peut avoir des effets physiologiques spécifiques (Calebiro et al., 2009, 2010 ; Ferrandon et al., 2009 ; Mullershausen et al., 2009 ; Feinstein et al., 2011, 2013 ; Irannejad et al., 2013 ; Vilardaga et al., 2014 ; Tsvetanova et al., 2015 ; Ismail et al., 2016). Ces découvertes sont en contradiction avec la vision classique de la signalisation des RCPG dans laquelle une interaction persistante de la β-arrestine avec le récepteur devrait empêcher l’activation de la protéine G. La cristallographie aux rayons X du récepteur β2 adrénergique en complexe avec Gαs a révélé que l’interaction implique à la fois les domaines N-terminal et C-terminal de la sous-unité Gαs et le noyau du récepteur (c’est-à-dire la boucle intracellulaire 2 et les domaines transmembranaires 5 et 6) (Rasmussen et al., 2011). Une étude récente a révélé que la β-arrestine interagit avec deux sites différents sur le récepteur, l’un à l’extrémité carboxyle du récepteur phosphorylé et l’autre, au cœur du récepteur (Shukla et al., 2014). Il est important de noter que des complexes de récepteurs internalisés appelés « méga-complexes » composés d’un seul RCPG, d’une β-arrestine et d’une protéine G ont été récemment découverts et leur architecture et leur fonctionnalité décrites (Thomsen et al., 2016). Ceux-ci semblent se former préférentiellement avec des récepteurs qui interagissent fortement avec les β-arrestines via une extrémité carboxyle contenant des clusters de phosphorylation sérine/thréonine. En effet, l’analyse par cryo-EM de méga-complexes purifiés a révélé qu’un seul récepteur peut simultanément se lier à travers son noyau transmembranaire avec la protéine G et à travers son extrémité carboxyle phosphorylée avec la β-arrestine (Figure 1).
Une autre illustration très intéressante de l’importance des β-arrestines dans le contrôle des signaux dans l’espace et dans le temps provient d’une étude de la signalisation de l’AMPc par le récepteur de la relaxine RXFP1 (Halls & Cooper, 2010). La relaxine est connue pour circuler à une concentration très faible et est pourtant capable de déclencher la signalisation de l’AMPc (Halls, 2012). Un mécanisme moléculaire a été proposé qui implique l’assemblage constitutif d’un signalosome au RXFP1. Ce dernier est composé de Gαs, de Gβγ et de l’adénylyl cyclase 2 (AC2) couplée fonctionnellement à AKAP79, cette dernière étant liée à l’hélice 8 du RXFP1. De plus, il est important de noter que la β-arrestine 2 s’associe simultanément à l’extrémité carboxyle de RXFP1 et échafaude la protéine kinase A (PKA) et la PDE4D3. Dans ce signalosome, l’activation de l’AC2 est donc toniquement contenue par la PDE4D3 activée par la PKA. Ce signalosome RXFP1 permet au récepteur de répondre à des concentrations attomolaires de relaxine (Halls & Cooper, 2010). D’autres hormones – comme les gonadotrophines par exemple – activent également la signalisation de l’AMPc à très faible concentration circulante (i.e., EC50 dans la gamme picomolaire), ce qui est difficile à expliquer sur la seule base de l’occupation des récepteurs (Ayoub et al., 2015). L’existence de signalosomes pré-assemblés du même type que ceux décrits pour RXFP1 est donc potentiellement intéressante et mérite des investigations à l’avenir.
Récemment, deux articles ont apporté un nouvel éclairage sur les rôles distincts du domaine carboxyle phosphorylé et du noyau du récepteur dans la médiation de l’interaction de la β-arrestine et de l’activation par les RCPG (Eichel et al., 2018 ; Latorraca et al., 2018). Comme discuté plus haut, des études structurelles récentes de l’interaction de l’arrestine avec la rhodopsine dans la rétine et de celle de la β-arrestine avec d’autres RCPG ont révélé deux types d’interactions au sein des complexes récepteur-arrestine : l’une impliquant la queue cytoplasmique phosphorylée du RCPG et l’autre impliquant son cœur transmembranaire ou noyau (Shukla et al., 2014 ; Kang et al., 2015). Il a été suggéré que ces deux modes d’interaction fassent intervenir deux sites distincts des arrestines : un « capteur de phosphorylation » et un « capteur d’activation ». Utilisant des simulations moléculaires dynamiques de l’arrestine au niveau atomique, Latorraca et al. (2018) ont étudié les mécanismes structurels par lesquels la queue cytoplasmique phosphorylée et le noyau transmembranaire régulent l’activation de l’arrestine. Les auteurs montrent que le déplacement de sa queue carboxyle conduit à une prolongation de l’état actif de l’arrestine, même lorsqu’elle n’est pas liée au récepteur. Cela peut expliquer le fait que la β-arrestine reste active et continue de « signaler » même après sa dissociation du RCPG activé (Eichel et al., 2016). En utilisant la structure rhodopsine-arrestine comme modèle pour leur simulation, les auteurs démontrent que le noyau du récepteur et la queue carboxyle stabilisent chacun individuellement les conformations actives de l’arrestine. La liaison simultanée des deux régions ajoute une stabilisation supplémentaire. De façon intéressante, les résultats de Latorraca et al. (2018), obtenus principalement par des simulations moléculaires dynamiques, sont cohérents avec ceux d’Eichel et al. (2018) qui, à l’aide de diverses techniques de biologie cellulaire, démontrent également que les interactions transitoires du cœur transmembranaire du RCPG sont suffisantes pour activer la β-arrestine. Eichel et al. (2018) montrent que l’activation et la signalisation soutenues de la β-arrestine après sa dissociation du RCPG nécessitent une interaction uniquement avec le cœur transmembranaire et pas avec la queue cytoplasmique. Cette activation de la β-arrestine dans la membrane plasmique implique une série d’interactions avec les phosphoinositides membranaires, et son accumulation dans les puits à clathrine (Figure 2).
Une telle activation multimodale des arrestines par le biais d’interactions avec le cœur transmembranaire du RCPG ou avec sa queue cytoplasmique souligne la chorégraphie complexe qui sous-tend les diverses fonctions régulatrices des β-arrestines.
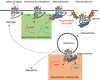 |
Figure 1 Représentation schématique du rôle des β-arrestines dans les différentes phases de l’activation et du trafic des RCPG. La signalisation est initiée depuis la membrane plasmique puis rapidement désensibilisée. Une seconde phase de signalisation plus persistante est alors déclenchée par la formation d’un méga-complexe dans l’endosome. PDE : phosphodiestérase ; GRK : kinases spécifiques des RCPG. |
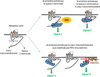 |
Figure 2 Représentation schématique des différents mécanismes d’activation de la signalisation par les β-arrestines. Les β-arrestines peuvent induire des signaux intracellulaires soit en s’associant au RCPG phosphorylé via sa queue C-terminale ou via sa queue C-terminale et son cœur transmembranaire (mécanisme classique, en haut), soit en s’associant transitoirement au RCPG non phosphorylé via son cœur transmembranaire, ce qui induit l’association de phosphatidylinositol biphosphate (PIP2) avec les β-arrestines, stabilisant ainsi la conformation active de ces dernières qui peuvent alors induire des signaux depuis des structures membranaires contenant la clathrine (mécanisme émergent, en bas). L’existence même et/ou la proportion entre ces différents modes d’activation varient en fonction du récepteur et du contexte cellulaire. |
Conclusions
Les β-arrestines sont reconnues comme des acteurs majeurs de la fonction des RCPG depuis près de trois décennies. La mise en évidence de leur implication dans le phénomène de biais pharmacologiques a ouvert la possibilité d’activer ou d’inhiber, tout ou partie de leurs fonctions, grâce à des ligands sélectifs de RCPG. L’intérêt thérapeutique de certains ligands biaisés de ce type est maintenant avéré. Ces avancées, conjuguées à la meilleure compréhensions que nous avons maintenant des mécanismes moléculaires et cellulaires utilisés par les β-arrestines pour induire des réponses biologiques, ouvrent la voie au développement rationnel de médicaments capables de contrôler les contributions relatives des protéines G et celles des β-arrestines en aval de RCPG cibles. De tels médicaments présenteront potentiellement une meilleure fenêtre thérapeutique et sont donc porteurs d’innovation et d’espoir.
Références
- Ahn, S., Kim, J., Hara, M.R., Ren, X.R., Lefkowitz, R.J. (2009). {beta}-arrestin 2 mediates anti-apoptotic signaling through regulation of BAD phosphorylation. J Biol Chem, 284, 8855-8865. [CrossRef] [PubMed] [Google Scholar]
- Ahn, S., Shenoy, S.K., Luttrell, L.M., Lefkowitz, R.J. (2020). SnapShot: beta-arrestin functions. Cell, 182, 1362-1362.e1. [CrossRef] [PubMed] [Google Scholar]
- Ayoub, M.A., Landomiel, F., Gallay, N., Jegot, G., Poupon, A., Crépieux, P., Reiter, E. (2015). Assessing gonadotropin receptor function by resonance energy transfer-based assays. Front Endocrinol (Lausanne), 6, 130. [CrossRef] [PubMed] [Google Scholar]
- Barnes, W.G., Reiter, E., Violin, J.D., Ren, X.R., Milligan, G., Lefkowitz, R.J. (2005). Beta-arrestin 1 and galphaq/11 coordinately activate RhoA and stress fiber formation following receptor stimulation. J Biol Chem, 280, 8041-8050. [CrossRef] [PubMed] [Google Scholar]
- Beaulieu, J.M., Sotnikova, T.D., Marion, S., Lefkowitz, R.J., Gainetdinov, R.R., Caron, M.G. (2005). An akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell, 122, 261-273. [CrossRef] [PubMed] [Google Scholar]
- Bruchas, M.R., Macey, T.A., Lowe, J.D., Chavkin, C. (2006). Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J Biol Chem, 281, 18081-18089. [CrossRef] [PubMed] [Google Scholar]
- Busillo, J.M., Armando, S., Sengupta, R., Meucci, O., Bouvier, M., Benovic, J.L. (2010). Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J Biol Chem, 285, 7805-7817. [CrossRef] [PubMed] [Google Scholar]
- Butcher, A.J., Prihandoko, R., Kong, K.C., Mcwilliams, P., Edwards, J.M., Bottrill, A., Mistry, S., Tobin, A.B. (2011). Differential G-protein-coupled receptor phosphorylation provides evidence for a signaling bar code. J Biol Chem, 286, 11506-11518. [CrossRef] [PubMed] [Google Scholar]
- Calebiro, D., Nikolaev, V.O., Gagliani, M.C., De Filippis, T., Dees, C., Tacchetti, C., Persani, L., Lohse, M.J. (2009). Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol, 7, e1000172. [CrossRef] [PubMed] [Google Scholar]
- Calebiro, D., Nikolaev, V.O., Persani, L., Lohse, M.J. (2010). Signaling by internalized G-protein-coupled receptors. Trends Pharmacol Sci, 31, 221-228. [CrossRef] [PubMed] [Google Scholar]
- Carr, R. 3rd, Benovic, J.L. (2016). From biased signalling to polypharmacology: Unlocking unique intracellular signalling using pepducins. Biochem Soc Trans, 44, 555-561. [CrossRef] [PubMed] [Google Scholar]
- Cassier, E., Gallay, N., Bourquard, T., Claeysen, S., Bockaert, J., Crépieux, P., Poupon, A., Reiter, E., Marin, P., Vandermoere, F. (2017). Phosphorylation of beta-arrestin2 at Thr(383) by MEK underlies beta-arrestin-dependent activation of Erk1/2 by GPCRs. Elife, 6. [CrossRef] [Google Scholar]
- Changeux, J.P., Christopoulos, A. (2016). Allosteric modulation as a unifying mechanism for receptor function and regulation. Cell, 166, 1084-1102. [CrossRef] [PubMed] [Google Scholar]
- Charest, P.G., Terrillon, S., Bouvier, M. (2005). Monitoring agonist-promoted conformational changes of beta-arrestin in living cells by intramolecular BRET. EMBO Rep, 6, 334-340. [CrossRef] [PubMed] [Google Scholar]
- Claing, A., Chen, W., Miller, W.E., Vitale, N., Moss, J., Prémont, R.T., Lefkowitz, R.J. (2001). beta-arrestin-mediated ADP-ribosylation factor 6 activation and beta 2-adrenergic receptor endocytosis. J Biol Chem, 276, 42509-42513. [CrossRef] [PubMed] [Google Scholar]
- De Lean, A., Stadel, J.M., Lefkowitz, R.J. (1980). A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem, 255, 7108-7117. [CrossRef] [PubMed] [Google Scholar]
- DeVree, B.T., Mahoney, J.P., Velez-Ruiz, G.A., Rasmussen, S.G., Kuszak, A.J., Edwald, E., Fung, J.J., Manglik, A., Masureel, M., Du, Y., Matt, R.A., Pardon, E., Steyaert, J., Kobilka, B.K., Sunahara, R.K. (2016). Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature, 535, 182-186. [CrossRef] [PubMed] [Google Scholar]
- DeWire, S.M., Violin, J.D. (2011). Biased ligands for better cardiovascular drugs: dissecting G-protein-coupled receptor pharmacology. Circ Res, 109, 205-216. [CrossRef] [PubMed] [Google Scholar]
- DeWire, S.M., Ahn, S., Lefkowitz, R.J., Shenoy, S.K. (2007). Beta-arrestins and cell signaling. Annu Rev Physiol, 69, 483-510. [CrossRef] [PubMed] [Google Scholar]
- DeWire, S.M., Kim, J., Whalen, E.J., Ahn, S., Chen, M., Lefkowitz, R.J. (2008). Beta-arrestin-mediated signaling regulates protein synthesis. J Biol Chem, 283, 10611-10620. [CrossRef] [PubMed] [Google Scholar]
- Eichel, K., Jullie, D., von Zastrow, M. (2016). beta-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat Cell Biol, 18, 303-310. [CrossRef] [PubMed] [Google Scholar]
- Eichel, K., Jullie, D., Barsi-Rhyne, B., Latorraca, N.R., Masureel, M., Sibarita, J.B., Dror, R.O., von Zastrow, M. (2018). Catalytic activation of beta-arrestin by GPCRs. Nature, 557, 381-386. [CrossRef] [PubMed] [Google Scholar]
- Feinstein, T.N., Wehbi, V.L., Ardura, J.A., Wheeler, D.S., Ferrandon, S., Gardella, T.J., Vilardaga, J.P. (2011). Retromer terminates the generation of cAMP by internalized PTH receptors. Nat Chem Biol, 7, 278-284. [CrossRef] [PubMed] [Google Scholar]
- Feinstein, T.N., Yui, N., Webber, M.J., Wehbi, V.L., Stevenson, H.P., King, J.D., Jr., Hallows, K.R., Brown, D., Bouley, R., Vilardaga, J.P. (2013). Noncanonical control of vasopressin receptor type 2 signaling by retromer and arrestin. J Biol Chem, 288, 27849-27860. [CrossRef] [PubMed] [Google Scholar]
- Ferrandon, S., Feinstein, T.N., Castro, M., Wang, B., Bouley, R., Potts, J.T., Gardella, T.J., Vilardaga, J.P. (2009). Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol, 5, 734-742. [Google Scholar]
- Freedman, N.J., Lefkowitz, R.J. (1996). Desensitization of G protein-coupled receptors. Recent Prog Horm Res, 51, 319-351; discussion 352-353. [PubMed] [Google Scholar]
- Galandrin, S., Oligny-Longpre, G., Bouvier, M. (2007). The evasive nature of drug efficacy: Implications for drug discovery. Trends Pharmacol Sci, 28, 423-430. [CrossRef] [PubMed] [Google Scholar]
- Gao, H., Sun, Y., Wu, Y., Luan, B., Wang, Y., Qu, B., Pei, G. (2004). Identification of beta-arrestin 2 as a G protein-coupled receptor-stimulated regulator of NF-kappaB pathways. Mol Cell, 14, 303-317. [CrossRef] [PubMed] [Google Scholar]
- Goodman, O.B., Jr., Krupnick, J.G., Santini, F., Gurevich, V.V., Penn, R.B., Gagnon, A.W., Keen, J.H., Benovic, J.L. (1996). Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature, 383, 447-450. [CrossRef] [PubMed] [Google Scholar]
- Gould, N., Doulias, P.T., Tenopoulou, M., Raju, K., Ischiropoulos, H. (2013). Regulation of protein function and signaling by reversible cysteine S-nitrosylation. J Biol Chem, 288, 26473-26479. [CrossRef] [PubMed] [Google Scholar]
- Grundmann, M., Merten, N., Malfacini, D., Inoue, A., Preis, P., Simon, K., Ruttiger, N., Ziegler, N., Benkel, T., Schmitt, N.K., Ishida, S., Muller, I., Reher, R., Kawakami, K., Inoue, A., Rick, U., Kuhl, T., Imhof, D., Aoki, J., Konig, G.M., Hoffmann, C., Gomeza, J., Wess, J., Kostenis, E. (2018). Lack of beta-arrestin signaling in the absence of active G proteins. Nat Commun, 9, 341. [CrossRef] [PubMed] [Google Scholar]
- Gurevich, V.V., Benovic, J.L. (1993). Visual arrestin interaction with rhodopsin. Sequential multisite binding ensures strict selectivity toward light-activated phosphorylated rhodopsin. J Biol Chem, 268, 11628-11638. [CrossRef] [PubMed] [Google Scholar]
- Halls, M.L. (2012). Constitutive formation of an RXFP1-signalosome: a novel paradigm in GPCR function and regulation. Br J Pharmacol, 165, 1644-1658. [CrossRef] [PubMed] [Google Scholar]
- Halls, M.L., Cooper, D.M. (2010). Sub-picomolar relaxin signalling by a pre-assembled RXFP1, AKAP79, AC2, beta-arrestin 2, PDE4D3 complex. EMBO J, 29, 2772-2787. [CrossRef] [PubMed] [Google Scholar]
- Heitzler, D., Durand, G., Gallay, N., Rizk, A., Ahn, S., Kim, J., Violin, J.D., Dupuy, L., Gauthier, C., Piketty, V., Crépieux, P., Poupon, A., Clément, F., Fages, F., Lefkowitz, R.J., Reiter, E. (2012). Competing G protein-coupled receptor kinases balance G protein and beta-arrestin signaling. Mol Syst Biol, 8, 590. [CrossRef] [PubMed] [Google Scholar]
- Irannejad, R., Tomshine, J.C., Tomshine, J.R., Chevalier, M., Mahoney, J.P., Steyaert, J., Rasmussen, S.G., Sunahara, R.K., El-Samad, H., Huang, B., von Zastrow, M. (2013). Conformational biosensors reveal GPCR signalling from endosomes. Nature, 495, 534-538. [CrossRef] [PubMed] [Google Scholar]
- Ismail, S., Gherardi, M.J., Froese, A., Zanoun, M., Gigoux, V., Clerc, P., Gaits-Iacovoni, F., Steyaert, J., Nikolaev, V.O., Fourmy, D. (2016). Internalized receptor for glucose-dependent insulinotropic peptide stimulates adenylyl cyclase on early endosomes. Biochem Pharmacol, 120, 33-45. [CrossRef] [PubMed] [Google Scholar]
- Iwata, K., Luo, J., Penn, R.B., Benovic, J.L. (2005). Bimodal regulation of the human H1 histamine receptor by G protein-coupled receptor kinase 2. J Biol Chem, 280, 2197-2204. [CrossRef] [PubMed] [Google Scholar]
- Jensen, J.B., Lyssand, J.S., Hague, C., Hille, B. (2009). Fluorescence changes reveal kinetic steps of muscarinic receptor-mediated modulation of phosphoinositides and Kv7.2/7.3 K+ channels. J Gen Physiol, 133, 347-359. [CrossRef] [PubMed] [Google Scholar]
- Kahsai, A.W., Wisler, J.W., Lee, J., Ahn, S., Cahill Iii, T.J., Dennison, S.M., Staus, D.P., Thomsen, A.R., Anasti, K.M., Pani, B., Wingler, L.M., Desai, H., Bompiani, K.M., Strachan, R.T., Qin, X., Alam, S.M., Sullenger, B.A., Lefkowitz, R.J. (2016). Conformationally selective RNA aptamers allosterically modulate the beta2-adrenoceptor. Nat Chem Biol, 12, 709-716. [CrossRef] [PubMed] [Google Scholar]
- Kang, J., Shi, Y., Xiang, B., Qu, B., Su, W., Zhu, M., Zhang, M., Bao, G., Wang, F., Zhang, X., Yang, R., Fan, F., Chen, X., Pei, G., Ma, L. (2005). A nuclear function of beta-arrestin1 in GPCR signaling: regulation of histone acetylation and gene transcription. Cell, 123, 833-847. [CrossRef] [PubMed] [Google Scholar]
- Kang, Y., Zhou, X.E., Gao, X., He, Y., Liu, W., Ishchenko, A., Barty, A., White, T.A., Yefanov, O., Han, G.W., Xu, Q., De Waal, P.W., Ke, J., Tan, M.H., Zhang, C., Moeller, A., West, G.M., Pascal, B.D., Van Eps, N., Caro, L.N., Vishnivetskiy, S.A., Lee, R.J., Suino-Powell, K.M., Gu, X., Pal, K., Ma, J., Zhi, X., Boutet, S., Williams, G.J., Messerschmidt, M., Gati, C., Zatsepin, N.A., Wang, D., James, D., Basu, S., Roy-Chowdhury, S., Conrad, C.E., Coe, J., Liu, H., Lisova, S., Kupitz, C., Grotjohann, I., Fromme, R., Jiang, Y., Tan, M., Yang, H., Li, J., Wang, M., Zheng, Z., Li, D., Howe, N., Zhao, Y., Standfuss, J., Diederichs, K., Dong, Y., Potter, C.S., Carragher, B., Caffrey, M., Jiang, H., Chapman, H.N., Spence, J.C., Fromme, P., Weierstall, U., Ernst, O.P., Katritch, V., Gurevich, V.V., Griffin, P.R., Hubbell, W.L., Stevens, R.C., Cherezov, V., Melcher, K., Xu, H.E. (2015). Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature, 523, 561-567. [CrossRef] [PubMed] [Google Scholar]
- Kara, E., Crépieux, P., Gauthier, C., Martinat, N., Piketty, V., Guillou, F., Reiter, E. (2006). A phosphorylation cluster of five serine and threonine residues in the C-terminus of the follicle-stimulating hormone receptor is important for desensitization but not for beta-arrestin-mediated ERK activation. Mol Endocrinol, 20, 3014-3026. [CrossRef] [PubMed] [Google Scholar]
- Kenakin, T. (2003). Ligand-selective receptor conformations revisited: the promise and the problem. Trends Pharmacol Sci, 24, 346-354. [CrossRef] [PubMed] [Google Scholar]
- Khoury, E., Nikolajev, L., Simaan, M., Namkung, Y., Laporte, S.A. (2014). Differential regulation of endosomal GPCR/beta-arrestin complexes and trafficking by MAPK. J Biol Chem, 289, 23302-23317. [CrossRef] [PubMed] [Google Scholar]
- Kim, Y.M., Barak, L.S., Caron, M.G., Benovic, J.L. (2002). Regulation of arrestin-3 phosphorylation by casein kinase II. J Biol Chem, 277, 16837-16846. [CrossRef] [PubMed] [Google Scholar]
- Kim, J., Ahn, S., Ren, X.R., Whalen, E.J., Reiter, E., Wei, H., Lefkowitz, R.J. (2005). Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proc Natl Acad Sci USA, 102, 1442-1447. [CrossRef] [PubMed] [Google Scholar]
- Kobilka, B.K. (2011). Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol Sci, 32, 213-218. [CrossRef] [PubMed] [Google Scholar]
- Kook, S., Zhan, X., Kaoud, T.S., Dalby, K.N., Gurevich, V.V., Gurevich, E.V. (2013). Arrestin-3 binds c-Jun N-terminal kinase 1 (JNK1) and JNK2 and facilitates the activation of these ubiquitous JNK isoforms in cells via scaffolding. J Biol Chem, 288, 37332-37342. [CrossRef] [PubMed] [Google Scholar]
- Landomiel, F., Gallay, N., Jegot, G., Tranchant, T., Durand, G., Bourquard, T., Crépieux, P., Poupon, A., Reiter, E. (2014). Biased signalling in follicle stimulating hormone action. Mol Cell Endocrinol, 382, 452-459. [CrossRef] [PubMed] [Google Scholar]
- Laporte, S.A., Oakley, R.H., Zhang, J., Holt, J.A., Ferguson, S.S., Caron, M.G., Barak, L.S. (1999). The beta2-adrenergic receptor/betaarrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Natl Acad Sci USA, 96, 3712-3717. [CrossRef] [PubMed] [Google Scholar]
- Latorraca, N.R., Wang, J.K., Bauer, B., Townshend, R.J.L., Hollingsworth, S.A., Olivieri, J.E., Xu, H.E., Sommer, M.E., Dror, R.O. (2018). Molecular mechanism of GPCR-mediated arrestin activation. Nature, 557, 452-456. [CrossRef] [PubMed] [Google Scholar]
- Lee, M.H., Appleton, K.M., Strungs, E.G., Kwon, J.Y., Morinelli, T.A., Peterson, Y.K., Laporte, S.A., Luttrell, L.M. (2016). The conformational signature of beta-arrestin2 predicts its trafficking and signalling functions. Nature, 531, 665-668. [CrossRef] [PubMed] [Google Scholar]
- Lefkowitz, R.J., Shenoy, S.K. (2005). Transduction of receptor signals by beta-arrestins. Science, 308, 512-517. [CrossRef] [PubMed] [Google Scholar]
- Lima-Fernandes, E., Enslen, H., Camand, E., Kotelevets, L., Boularan, C., Achour, L., Benmerah, A., Gibson, L.C., Baillie, G.S., Pitcher, J.A., Chastre, E., Etienne-Manneville, S., Marullo, S., Scott, M.G. (2011). Distinct functional outputs of PTEN signalling are controlled by dynamic association with beta-arrestins. EMBO J, 30, 2557-2568. [CrossRef] [PubMed] [Google Scholar]
- Lin, F.T., Krueger, K.M., Kendall, H.E., Daaka, Y., Fredericks, Z.L., Pitcher, J.A., Lefkowitz, R.J. (1997). Clathrin-mediated endocytosis of the beta-adrenergic receptor is regulated by phosphorylation/dephosphorylation of beta-arrestin1. J Biol Chem, 272, 31051-31057. [CrossRef] [PubMed] [Google Scholar]
- Lin, F.T., Miller, W.E., Luttrell, L.M., Lefkowitz, R.J. (1999). Feedback regulation of beta-arrestin 1 function by extracellular signal-regulated kinases. J Biol Chem, 274, 15971-15974. [CrossRef] [PubMed] [Google Scholar]
- Lin, F.T., Chen, W., Shenoy, S., Cong, M., Exum, S.T., Lefkowitz, R.J. (2002). Phosphorylation of beta-arrestin2 regulates its function in internalization of beta(2)-adrenergic receptors. Biochemistry, 41, 10692-10699. [CrossRef] [PubMed] [Google Scholar]
- Lohse, M.J., Calebiro, D. (2013). Cell biology: Receptor signals come in waves. Nature, 495, 457-458. [CrossRef] [PubMed] [Google Scholar]
- Lohse, M.J., Hofmann, K.P. (2015). Spatial and temporal aspects of signaling by G protein-coupled receptors. Mol Pharmacol, 88, 572-578. [CrossRef] [PubMed] [Google Scholar]
- Lohse, M.J., Nikolaev, V.O., Hein, P., Hoffmann, C., Vilardaga, J.P., Bunemann, M. (2008). Optical techniques to analyze real-time activation and signaling of G-protein-coupled receptors. Trends Pharmacol Sci, 29, 159-165. [CrossRef] [MathSciNet] [PubMed] [Google Scholar]
- Luo, J., Busillo, J.M., Benovic, J.L. (2008). M3 muscarinic acetylcholine receptor-mediated signaling is regulated by distinct mechanisms. Mol Pharmacol, 74, 338-347. [CrossRef] [PubMed] [Google Scholar]
- Luttrell, L.M., Roudabush, F.L., Choy, E.W., Miller, W.E., Field, M.E., Pierce, K.L., Lefkowitz, R.J. (2001). Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci USA, 98, 2449-2454. [CrossRef] [PubMed] [Google Scholar]
- Luttrell, L.M., Wang, J., Plouffe, B., Smith, J.S., Yamani, L., Kaur, S., Jean-Charles, P.Y., Gauthier, C., Lee, M.H., Pani, B., Kim, J., Ahn, S., Rajagopal, S., Reiter, E., Bouvier, M., Shenoy, S.K., Laporte, S.A., Rockman, H.A., Lefkowitz, R.J. (2018). Manifold roles of beta-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci Signal, 11(549), eaat7650. [CrossRef] [PubMed] [Google Scholar]
- Martini, L., Hastrup, H., Holst, B., Fraile-Ramos, A., Marsh, M., Schwartz, T.W. (2002). NK1 receptor fused to beta-arrestin displays a single-component, high-affinity molecular phenotype. Mol Pharmacol, 62, 30-37. [CrossRef] [PubMed] [Google Scholar]
- McDonald, P.H., Cote, N.L., Lin, F.T., Prémont, R.T., Pitcher, J.A., Lefkowitz, R.J. (1999). Identification of NSF as a beta-arrestin1-binding protein. Implications for beta2-adrenergic receptor regulation. J Biol Chem, 274, 10677-10680. [CrossRef] [PubMed] [Google Scholar]
- McDonald, P.H., Chow, C.W., Miller, W.E., Laporte, S.A., Field, M.E., Lin, F.T., Davis, R.J., Lefkowitz, R.J. (2000). Beta-arrestin 2: A receptor-regulated MAPK scaffold for the activation of JNK3. Science, 290, 1574-1577. [CrossRef] [PubMed] [Google Scholar]
- Mujic-Delic, A., De Wit, R.H., Verkaar, F., Smit, M.J. (2014). GPCR-targeting nanobodies: attractive research tools, diagnostics, and therapeutics. Trends Pharmacol Sci, 35, 247-255. [CrossRef] [PubMed] [Google Scholar]
- Mullershausen, F., Zecri, F., Cetin, C., Billich, A., Guerini, D., Seuwen, K. (2009). Persistent signaling induced by FTY720-phosphate is mediated by internalized S1P1 receptors. Nat Chem Biol, 5, 428-434. [Google Scholar]
- Nelson, C.D., Perry, S.J., Regier, D.S., Prescott, S.M., Topham, M.K., Lefkowitz, R.J. (2007). Targeting of diacylglycerol degradation to M1 muscarinic receptors by beta-arrestins. Science, 315, 663-666. [CrossRef] [PubMed] [Google Scholar]
- Nobles, K.N., Guan, Z., Xiao, K., Oas, T.G., Lefkowitz, R.J. (2007). The active conformation of beta-arrestin1: direct evidence for the phosphate sensor in the N-domain and conformational differences in the active states of beta-arrestins 1 and 2. J Biol Chem, 282, 21370-21381. [CrossRef] [PubMed] [Google Scholar]
- Nobles, K.N., Xiao, K., Ahn, S., Shukla, A.K., Lam, C.M., Rajagopal, S., Strachan, R.T., Huang, T.Y., Bressler, E.A., Hara, M.R., Shenoy, S.K., Gygi, S.P., Lefkowitz, R.J. (2011). Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci Signal, 4, ra51. [CrossRef] [PubMed] [Google Scholar]
- Noma, T., Lemaire, A., Naga Prasad, S.V., Barki-Harrington, L., Tilley, D.G., Chen, J., Le Corvoisier, P., Violin, J.D., Wei, H., Lefkowitz, R.J., Rockman, H.A. (2007). Beta-arrestin-mediated beta1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J Clin Invest, 117, 2445-2458. [Google Scholar]
- Nuber, S., Zabel, U., Lorenz, K., Nuber, A., Milligan, G., Tobin, A.B., Lohse, M.J., Hoffmann, C. (2016). beta-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature, 531, 661-664. [CrossRef] [PubMed] [Google Scholar]
- Nygaard, R., Zou, Y., Dror, R.O., Mildorf, T.J., Arlow, D.H., Manglik, A., Pan, A.C., Liu, C.W., Fung, J.J., Bokoch, M.P., Thian, F.S., Kobilka, T.S., Shaw, D.E., Mueller, L., Prosser, R.S., Kobilka, B.K. (2013). The dynamic process of beta(2)-adrenergic receptor activation. Cell, 152, 532-542. [CrossRef] [PubMed] [Google Scholar]
- O’Hayre, M., Eichel, K., Avino, S., Zhao, X., Steffen, D.J., Feng, X., Kawakami, K., Aoki, J., Messer, K., Sunahara, R., Inoue, A., Von Zastrow, M., Gutkind, J.S. (2017). Genetic evidence that beta-arrestins are dispensable for the initiation of beta2-adrenergic receptor signaling to ERK. Sci Signal, 10(484), eaal 3395. [Google Scholar]
- Oakley, R.H., Laporte, S.A., Holt, J.A., Caron, M.G., Barak, L.S. (2000). Differential affinities of visual arrestin, beta arrestin1, and beta arrestin 2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem, 275, 17201-17210. [CrossRef] [PubMed] [Google Scholar]
- Oakley, R.H., Laporte, S.A., Holt, J.A., Barak, L.S., Caron, M.G. (2001). Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-beta-arrestin complexes after receptor endocytosis. J Biol Chem, 276, 19452-19460. [CrossRef] [PubMed] [Google Scholar]
- Ozawa, K., Whalen, E.J., Nelson, C.D., Mu, Y., Hess, D.T., Lefkowitz, R.J., Stamler, J.S. (2008). S-nitrosylation of beta-arrestin regulates beta-adrenergic receptor trafficking. Mol Cell, 31, 395-405. [CrossRef] [MathSciNet] [PubMed] [Google Scholar]
- Perroy, J., Pontier, S., Charest, P.G., Aubry, M., Bouvier, M. (2004). Real-time monitoring of ubiquitination in living cells by BRET. Nat Methods, 1, 203-208. [CrossRef] [PubMed] [Google Scholar]
- Perry, S.J., Baillie, G.S., Kohout, T.A., McPhee, I., Magiera, M.M., Ang, K.L., Miller, W.E., McLean, A.J., Conti, M., Houslay, M.D., Lefkowitz, R.J. (2002). Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science, 298, 834-836. [CrossRef] [PubMed] [Google Scholar]
- Rasmussen, S.G., DeVree, B.T., Zou, Y., Kruse, A.C., Chung, K.Y., Kobilka, T.S., Thian, F.S., Chae, P.S., Pardon, E., Calinski, D., Mathiesen, J.M., Shah, S.T., Lyons, J.A., Caffrey, M., Gellman, S.H., Steyaert, J., Skiniotis, G., Weis, W.I., Sunahara, R.K., Kobilka, B.K. (2011). Crystal structure of the beta 2 adrenergic receptor-Gs protein complex. Nature, 477, 549-555. [CrossRef] [PubMed] [Google Scholar]
- Reiter, E., Lefkowitz, R.J. (2006). GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab, 17, 159-165. [CrossRef] [PubMed] [Google Scholar]
- Reiter, E., Ahn, S., Shukla, A.K., Lefkowitz, R.J. (2012). Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol, 52, 179-197. [CrossRef] [PubMed] [Google Scholar]
- Ren, X.R., Reiter, E., Ahn, S., Kim, J., Chen, W., Lefkowitz, R.J. (2005). Different G protein-coupled receptor kinases govern G protein and beta-arrestin-mediated signaling of V2 vasopressin receptor. Proc Natl Acad Sci USA, 102, 1448-1453. [CrossRef] [PubMed] [Google Scholar]
- Shenoy, S.K., McDonald, P.H., Kohout, T.A., Lefkowitz, R.J. (2001). Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science, 294, 1307-1313. [CrossRef] [PubMed] [Google Scholar]
- Shenoy, S.K., Drake, M.T., Nelson, C.D., Houtz, D.A., Xiao, K., Madabushi, S., Reiter, E., Prémont, R.T., Lichtarge, O., Lefkowitz, R.J. (2006). β-arrestin-dependent, G protein-independent ERK1/2 activation by the beta 2 adrenergic receptor. J Biol Chem, 281, 1261-1273. [CrossRef] [PubMed] [Google Scholar]
- Shenoy, S.K., Barak, L.S., Xiao, K., Ahn, S., Berthouze, M., Shukla, A.K., Luttrell, L.M., Lefkowitz, R.J. (2007). Ubiquitination of beta-arrestin links seven-transmembrane receptor endocytosis and ERK activation. J Biol Chem, 282, 29549-29562. [CrossRef] [PubMed] [Google Scholar]
- Shukla, A.K., Violin, J.D., Whalen, E.J., Gesty-Palmer, D., Shenoy, S.K., Lefkowitz, R.J. (2008). Distinct conformational changes in beta-arrestin report biased agonism at seven-transmembrane receptors. Proc Natl Acad Sci USA, 105, 9988-9993. [CrossRef] [PubMed] [Google Scholar]
- Shukla, A.K., Westfield, G.H., Xiao, K., Reis, R.I., Huang, L.Y., Tripathi-Shukla, P., Qian, J., Li, S., Blanc, A., Oleskie, A.N., Dosey, A.M., Su, M., Liang, C.R., Gu, L.L., Shan, J.M., Chen, X., Hanna, R., Choi, M., Yao, X.J., Klink, B.U., Kahsai, A.W., Sidhu, S.S., Koide, S., Penczek, P.A., Kossiakoff, A.A., Woods, V.L., Jr., Kobilka, B.K., Skiniotis, G., Lefkowitz, R.J. (2014). Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature, 512, 218-222. [CrossRef] [PubMed] [Google Scholar]
- Staus, D.P., Wingler, L.M., Strachan, R.T., Rasmussen, S.G., Pardon, E., Ahn, S., Steyaert, J., Kobilka, B.K., Lefkowitz, R.J. (2014). Regulation of beta2-adrenergic receptor function by conformationally selective single-domain intrabodies. Mol Pharmacol, 85, 472-481 [CrossRef] [PubMed] [Google Scholar]
- Staus, D.P., Strachan, R.T., Manglik, A., Pani, B., Kahsai, A.W., Kim, T.H., Wingler, L.M., Ahn, S., Chatterjee, A., Masoudi, A., Kruse, A.C., Pardon, E., Steyaert, J., Weis, W.I., Prosser, R.S., Kobilka, B.K., Costa, T., Lefkowitz, R.J. (2016). Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature, 535, 448-452. [CrossRef] [PubMed] [Google Scholar]
- Strachan, R.T., Sun, J.P., Rominger, D.H., Violin, J.D., Ahn, S., Rojas Bie Thomsen, A., Zhu, X., Kleist, A., Costa, T., Lefkowitz, R.J. (2014). Divergent transducer-specific molecular efficacies generate biased agonism at a G protein-coupled receptor (GPCR). J Biol Chem, 289, 14211-14224. [CrossRef] [PubMed] [Google Scholar]
- Sun, Y., Cheng, Z., Ma, L., Pei, G. (2002). Beta-arrestin 2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J Biol Chem, 277, 49212-49219. [CrossRef] [PubMed] [Google Scholar]
- Thomsen, A.R., Plouffe, B., Cahill, T.J. 3rd, Shukla, A.K., Tarrasch, J.T., Dosey, A.M., Kahsai, A.W., Strachan, R.T., Pani, B., Mahoney, J.P., Huang, L., Breton, B., Heydenreich, F.M., Sunahara, R.K., Skiniotis, G., Bouvier, M., Lefkowitz, R.J. (2016). GPCR-G protein-beta-arrestin super-complex mediates sustained G protein signaling. Cell, 166, 907-919. [CrossRef] [PubMed] [Google Scholar]
- Tobin, A.B., Butcher, A.J., Kong, K.C. (2008). Location, location, location..site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol Sci, 29, 413-420. [CrossRef] [PubMed] [Google Scholar]
- Tsvetanova, N.G., Irannejad, R., von Zastrow, M. (2015). G protein-coupled receptor (GPCR) signaling via heterotrimeric G proteins from endosomes. J Biol Chem, 290, 6689-6696. [CrossRef] [PubMed] [Google Scholar]
- Vilardaga, J.P., Jean-Alphonse, F.G., Gardella, T.J. (2014). Endosomal generation of cAMP in GPCR signaling. Nat Chem Biol, 10, 700-706. [Google Scholar]
- Violin, J.D., Lefkowitz, R.J. (2007). Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci, 28, 416-422. [CrossRef] [PubMed] [Google Scholar]
- Wacker, D., Wang, C., Katritch, V., Han, G.W., Huang, X.P., Vardy, E., McCorvy, J.D., Jiang, Y., Chu, M., Siu, F.Y., Liu, W., Xu, H.E., Cherezov, V., Roth, B.L., Stevens, R.C. (2013). Structural features for functional selectivity at serotonin receptors. Science, 340, 615-619. [CrossRef] [PubMed] [Google Scholar]
- Walters, R.W., Shukla, A.K., Kovacs, J.J., Violin, J.D., DeWire, S.M., Lam, C.M., Chen, J.R., Muehlbauer, M.J., Whalen, E.J., Lefkowitz, R.J. (2009). beta-Arrestin 1 mediates nicotinic acid-induced flushing, but not its antilipolytic effect, in mice. J Clin Invest, 119, 1312-1321. [CrossRef] [PubMed] [Google Scholar]
- Watson, C., Chen, G., Irving, P., Way, J., Chen, W.J., Kenakin, T. (2000). The use of stimulus-biased assay systems to detect agonist-specific receptor active states: implications for the trafficking of receptor stimulus by agonists. Mol Pharmacol, 58, 1230-1238. [CrossRef] [PubMed] [Google Scholar]
- Wei, H., Ahn, S., Shenoy, S.K., Karnik, S.S., Hunyady, L., Luttrell, L.M., Lefkowitz, R.J. (2003). Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci USA, 100, 10782-10787. [CrossRef] [PubMed] [Google Scholar]
- Whalen, E.J., Foster, M.W., Matsumoto, A., Ozawa, K., Violin, J.D., Que, L.G., Nelson, C.D., Benhar, M., Keys, J.R., Rockman, H.A., Koch, W.J., Daaka, Y., Lefkowitz, R.J., Stamler, J.S. (2007). Regulation of beta-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell, 129, 511-522. [CrossRef] [PubMed] [Google Scholar]
- Whalen, E.J., Rajagopal, S., Lefkowitz, R.J. (2011). Therapeutic potential of beta-arrestin- and G protein-biased agonists. Trends Mol Med, 17, 126-139. [CrossRef] [PubMed] [Google Scholar]
- Wyatt, D., Malik, R., Vesecky, A.C., Marchese, A. (2011). Small ubiquitin-like modifier modification of arrestin-3 regulates receptor trafficking. J Biol Chem, 286, 3884-3893. [CrossRef] [PubMed] [Google Scholar]
- Xiao, K., Shenoy, S.K., Nobles, K., Lefkowitz, R.J. (2004). Activation-dependent conformational changes in {beta}-arrestin 2. J Biol Chem, 279, 55744-55753. [CrossRef] [PubMed] [Google Scholar]
- Xiao, K., McClatchy, D.B., Shukla, A.K., Zhao, Y., Chen, M., Shenoy, S.K., Yates, J.R. 3rd, Lefkowitz, R.J. (2007). Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc Natl Acad USA, 104, 12011-12016. [CrossRef] [PubMed] [Google Scholar]
- Xiao, K., Sun, J., Kim, J., Rajagopal, S., Zhai, B., Villen, J., Haas, W., Kovacs, J.J., Shukla, A.K., Hara, M.R., Hernandez, M., Lachmann, A., Zhao, S., Lin, Y., Cheng, Y., Mizuno, K., Ma’ayan, A., Gygi, S.P., Lefkowitz, R.J. (2010). Global phosphorylation analysis of beta-arrestin-mediated signaling downstream of a seven transmembrane receptor (7TMR). Proc Natl Acad Sci USA, 107, 15299-15304. [CrossRef] [PubMed] [Google Scholar]
- Yan, D., Stocco, R., Sawyer, N., Nesheim, M.E., Abramovitz, M., Funk, C.D. (2011). Differential signaling of cysteinyl leukotrienes and a novel cysteinyl leukotriene receptor 2 (CysLT(2)) agonist, N-methyl-leukotriene C(4), in calcium reporter and beta arrestin assays. Mol Pharmacol, 79, 270-278. [CrossRef] [PubMed] [Google Scholar]
- Zidar, D.A., Violin, J.D., Whalen, E.J., Lefkowitz, R.J. (2009). Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc Natl Acad Sci USA, 106, 9649-9654. [CrossRef] [PubMed] [Google Scholar]
Citation de l’article: Reiter, E. (2021). Mécanismes d’action et rôles multiples des β-arrestines dans la biologie des récepteurs couplés aux protéines G. Biologie Aujourd’hui, 215, 107-118
Liste des figures
|
Figure 1 Représentation schématique du rôle des β-arrestines dans les différentes phases de l’activation et du trafic des RCPG. La signalisation est initiée depuis la membrane plasmique puis rapidement désensibilisée. Une seconde phase de signalisation plus persistante est alors déclenchée par la formation d’un méga-complexe dans l’endosome. PDE : phosphodiestérase ; GRK : kinases spécifiques des RCPG. |
| Dans le texte | |
|
Figure 2 Représentation schématique des différents mécanismes d’activation de la signalisation par les β-arrestines. Les β-arrestines peuvent induire des signaux intracellulaires soit en s’associant au RCPG phosphorylé via sa queue C-terminale ou via sa queue C-terminale et son cœur transmembranaire (mécanisme classique, en haut), soit en s’associant transitoirement au RCPG non phosphorylé via son cœur transmembranaire, ce qui induit l’association de phosphatidylinositol biphosphate (PIP2) avec les β-arrestines, stabilisant ainsi la conformation active de ces dernières qui peuvent alors induire des signaux depuis des structures membranaires contenant la clathrine (mécanisme émergent, en bas). L’existence même et/ou la proportion entre ces différents modes d’activation varient en fonction du récepteur et du contexte cellulaire. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.