| Numéro |
Biologie Aujourd’hui
Volume 215, Numéro 1-2, 2021
|
|
|---|---|---|
| Page(s) | 25 - 43 | |
| DOI | https://doi.org/10.1051/jbio/2021007 | |
| Publié en ligne | 16 août 2021 | |
Article
Dégradation induite des protéines par des molécules PROTAC et stratégies apparentées : développements à visée thérapeutique
Induced degradation of proteins by PROTACs and other strategies: towards promising drugs
Sorbonne Université, Institut de Biologie Paris Seine (IBPS), CNRS UMR 8256, Inserm ERL U1164,
7 quai Saint-Bernard,
75252
Paris Cedex 05, France
* Auteur correspondant : Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Reçu :
18
Juin
2021
Résumé
Alors que, pour la plupart, les médicaments actuels sont de petites molécules inhibant l’action d’une protéine en bloquant un site d’interaction, la dégradation ciblée des protéines, découverte il y a une vingtaine d’années via les petites molécules PROTAC, connaît aujourd’hui un très grand développement, aussi bien au niveau universitaire qu’industriel. Cette dégradation ciblée permet de contrôler la concentration intracellulaire d’une protéine spécifique comme peuvent le faire les techniques basées sur les acides nucléiques (oligonucléotides antisens, ARNsi, CRISPR-Cas9). Les molécules PROTAC sont des chimères hétéro-bifonctionnelles capables de lier simultanément une protéine spécifique devant être dégradée et une E3 ubiquitine ligase. Les PROTAC sont donc capables de provoquer l’ubiquitinylation de la protéine ciblée et sa dégradation par le protéasome 26S. De nature peptidique, puis non peptidique, les PROTAC sont maintenant administrables par voie orale. Ce détournement du système ubiquitine protéasome permet aux molécules PROTAC d’élargir considérablement le champ des applications thérapeutiques puisque l’élimination de protéines dépourvues de poches ou de crevasses bien définies, dites difficiles à cibler, devient possible. Cette technologie versatile a conduit à la dégradation d’une grande variété de protéines comme des facteurs de transcription, des sérine/thréonine/tyrosine kinases, des protéines de structure, des protéines cytosoliques, des lecteurs épigénétiques. Certaines ligases telles que VHL, MDM2, cereblon et IAP sont couramment utilisées pour être recrutées par les PROTAC. Actuellement, le nombre de ligases pouvant être utilisées ainsi que la nature des protéines dégradées sont en constante augmentation. Deux PROTAC sont en étude clinique pour les cancers du sein (ARV471) et de la prostate (ARV110). La dégradation spécifique d’une protéine par le protéasome peut aussi être induite par d’autres types de molécules synthétiques : colles moléculaires, marqueurs hydrophobes, HaloPROTAC, homo-PROTAC. D’autres constituants cellulaires sont aussi éligibles à une dégradation induite : ARN-PROTAC pour les protéines se liant à l’ARN et RIBOTAC pour la dégradation de l’ARN lui-même comme celui du SARS-CoV-2. Des dégradations induites en dehors du protéasome sont aussi connues : LYTAC, pour des chimères détournant la dégradation de protéines extracellulaires vers les lysosomes, et MADTAC, pour des chimères détournant la dégradation par macroautophagie. Plusieurs techniques, en particulier des plates-formes de criblage, la modélisation mathématique et la conception computationnelle sont utilisées pour le développement de nouveaux PROTAC efficaces.
Abstract
Targeted protein degradation (TPD), discovered twenty years ago through the PROTAC technology, is rapidly developing thanks to the implication of many scientists from industry and academia. PROTAC chimeras are heterobifunctional molecules able to link simultaneously a protein to be degraded and an E3 ubiquitin ligase. This allows the protein ubiquitination and its degradation by 26S proteasome. PROTACs have evolved from small peptide molecules to small non-peptide and orally available molecules. It was shown that PROTACs are capable to degrade proteins considered as “undruggable” i.e. devoid of well-defined pockets and deep grooves possibly occupied by small molecules. Among these “hard to drug” proteins, several can be degraded by PROTACs: scaffold proteins, BAF complex, transcription factors, Ras family proteins. Two PROTACs are clinically tested for breast (ARV471) and prostate (ARV110) cancers. The protein degradation by proteasome is also induced by other types of molecules: molecular glues, hydrophobic tagging (HyT), HaloPROTACs and homo-PROTACs. Other cellular constituents are eligible to induced degradation: RNA-PROTACs for RNA binding proteins and RIBOTACs for degradation of RNA itself (SARS-CoV-2 RNA). TPD has recently moved beyond the proteasome with LYTACs (lysosome targeting chimeras) and MADTACs (macroautophagy degradation targeting chimeras). Several techniques such as screening platforms together with mathematical modeling and computational design are now used to improve the discovery of new efficient PROTACs.
Mots clés : dégradation induite de protéines / protéasome / PROTAC / colles moléculaires / médicaments
Key words: targeted protein degradation / PROTACs / molecular glues / hydrophobic tagging / drugs
© Société de Biologie, 2021
Introduction
Les pathologies héritées ou acquises sont souvent liées au fonctionnement anormal d’une protéine qu’il convient de neutraliser (Figure 1A). Or, la majeure partie des protéines ne possèdent pas de ligands de petite taille connus et sont dépourvues de site actif ou d’une poche susceptible de les accommoder. Ceci constitue un obstacle très important dans la recherche de molécules bioactives pouvant conduire à des médicaments. Le mode d’action de la majorité des médicaments actuels est en effet basé sur l’occupation d’une poche de reconnaissance de la protéine ciblée par des petites molécules inhibitrices. Crews et al. parlent d’une pharmacologie pilotée par l’occupation temporaire de la protéine impliquée dans la pathologie (en dénomination anglaise, occupancy-driven pharmacology) (Figure 1A) (Petterson & Crews, 2019). La fraction de médicament liée à la protéine dépend directement de l’affinité du composé pour sa cible. Des ligands spécifiques et de forte affinité (si possible au niveau nanomolaire ou picomolaire) sont nécessaires pour avoir une activité biologique à des doses qui demeurent tolérables pour l’organisme. Plus l’inhibition est longue, plus le bénéfice clinique sera important. Ainsi des concentrations locales suffisamment élevées d’inhibiteurs doivent être maintenues pendant des temps longs pour assurer une efficacité thérapeutique importante (IC90-95), ce qui peut conduire à des effets secondaires et à des résistances puisque des protéines autres que la cible peuvent alors être atteintes (Adjei, 2006). De nouvelles classes de médicaments ont donc été activement recherchées pour éviter ces écueils. Une façon de procéder consiste à utiliser une molécule exogène qui déclenche un événement permettant de réduire l’abondance cellulaire de la protéine impliquée dans la pathologie (en dénomination anglaise, event-driven pharmacology) (Figures 1A et 1B) (Cromm & Crews, 2017). C’est le cas des thérapeutiques basées sur les acides nucléiques, les anticorps monoclonaux et les protéines recombinantes (Valeur et al., 2017). Des développements remarquables ont été conduits dans le domaine des ARN interférents (ARNsi), des oligonucléotides antisens et de l’édition du génome comme la stratégie CRISPR-Cas9 couronnée par le prix Nobel de Chimie 2020 décerné à Emmanuelle Charpentier et Jennifer Doudna (Cong et al., 2013). Cependant, l’utilisation d’acides nucléiques se trouve limitée par leur instabilité in vivo et leur faible biodisponibilité (Deng et al., 2014 ; Conde & Artzi, 2015). Une autre grande catégorie de stratégies utilise la dégradation induite et spécifique d’une protéine (Figure 1B). Parmi elles, la technologie PROTAC (PROteolysis Targeting Chimera) utilise un mode d’action lié à un évènement cellulaire (Toure & Crews, 2016 ; Cromm & Crews, 2017 ; Lai & Crews, 2017 ; Ottis & Crews, 2017 ; Salami & Crews, 2017 ; Bondeson et al., 2018 ; Burslem et al., 2018 ; Burslem & Crews, 2020). De petites molécules hétéro-bifonctionnelles sont utilisées pour capturer le système ubiquitine-protéasome afin d’induire la dégradation d’une cible protéique dite protéine d’intérêt. Leur effet va donc persister tant que de nouvelles molécules de la protéine ciblée ne sont pas biosynthétisées. Ces molécules exogènes, appelées essentiellement PROTAC, ont connu un développement spectaculaire à partir de 2017 (Figure 1C). La dégradation ciblée d’une protéine peut aussi être induite par d’autres types de molécules dont les colles moléculaires et des marqueurs hydrophobes qui induisent un repliement incorrect de la protéine. Enfin, l’induction d’une dégradation ciblée a été étendue à des constituants cellulaires autres que les protéines comme les ARN, les systèmes lysosome et macroautophagique pouvant être détournés par des chimères moléculaires.
La dernière décennie a donc vu un changement dans le domaine des cibles thérapeutiques pouvant être choisies pour développer des médicaments. On est passé de cibles traditionnelles comme, notamment, les récepteurs couplés aux protéines G, les enzymes et les canaux ioniques, à des cibles plus complexes à atteindre car réputées non « ciblables » en thérapeutique (undruggable en dénomination anglaise). Or de telles cibles peuvent être d’un grand intérêt pharmacologique (Valeur et al., 2017) bien que dépourvues de site susceptible d’accueillir un ligand qui déclencherait l’action à visée thérapeutique. Cet article présente les techniques développées pour induire de façon sélective à des fins thérapeutiques la dégradation d’une protéine à éliminer par détournement du système ubiquitine-protéasome (SUP). Leur évolution en tant qu’objets de recherche et objets thérapeutiques y est décrite et les applications à d’autres constituants cellulaires y sont également évoquées. Enfin, la dégradation induite des protéines par des molécules autres que les PROTAC fait l’objet d’un chapitre spécifique.
 |
Figure 1 Contrôle de l’action délétère d’une protéine impliquée dans une pathologie. A. Les deux modèles pharmacologiques classiques : interaction de la protéine avec un inhibiteur à forte concentration pour contrôler la fonction anormale ; contrôle de l’abondance de la protéine en diminuant son niveau intracellulaire par des technologies basées sur les acides nucléiques ou par des dégradations sélectives induites par des petites molécules. Alors que des petites molécules sont utilisées pour inhiber l’action d’une protéine en bloquant son site actif, les petites molécules PROTAC cooptent la machinerie cellulaire pour dégrader complètement la protéine cible. B. Résumé des techniques permettant le contrôle de l’abondance d’une protéine. C. Nombre de publications portant sur la technologie PROTAC au cours des dernières années. |
La dégradation des protéines par l’UPS et son contrôle
Les caractéristiques structurales, les rôles physiologiques et pathologiques des différents protéasomes sont décrits dans un article précédent (Reboud-Ravaux, 2021). On dégagera simplement ici les particularités de l’UPS ayant conduit à l’élaboration de ces nouvelles technologies d’induction sélective de dégradation d’une protéine.
Origine de la technologie. La technologie PROTAC est née de deux observations : (a) le détournement de l’UPS par les virus pour s’affranchir des défenses cellulaires (Mahon et al., 2014) ; (b) l’induction de la dégradation de facteurs de transcription par les hormones de plantes (Dharmasiri et al., 2005). En effet, un grand nombre de virus induisent la dégradation de protéines cellulaires pour contrecarrer les mécanismes de régulation et de défenses antivirales dont dispose la cellule. Ainsi, le virus de l’immunodéficience humaine (VIH-1) utilise quatre de ses protéines (VPU, VPR, VPX et VIF) pour cibler des protéines cellulaires (comme le CD4 et le complexe CMH 1 pour le VPU) et conduire à leur élimination via l’ubiquitinylation et la dégradation par le protéasome. Des virus à ADN codent des protéines qui peuvent se fixer et inactiver des facteurs cellulaires essentiels comme le suppresseur de tumeurs p53 (Dybas et al., 2018). Les relations UPS-virus influenza ont aussi été analysées (Karim et al., 2020) ainsi que celles impliquant le SARS-Cov-2. Une interaction du nouvel Orf10 du SARS-CoV-2 avec des membres d’un complexe Cullin 2 (CUL2) RING E3 ligase a été démontrée (Gordon et al., 2020).
Vie et mort des protéines. La dégradation sélective d’une protéine nécessite habituellement son ubiquitinylation ce qui permet son adressage vers le protéasome 26S (Figure 2). Une cascade séquentielle de trois types d’enzymes est impliquée dans l’attachement d’une courte chaîne de poly-ubiquitine : les enzymes d’activation de l’ubiquitine dépendant de l’ATP (E1), les enzymes de conjugaison à l’ubiquitine (E2) et les ubiquitine-protéine ligases E3. La particularité de ces dernières est une interaction spécifique avec une très grande variété de protéines-substrats. Cette étape de reconnaissance par les E3 est considérée comme l’étape lente de la dégradation des protéines. Il existe plus de 600 E3 dans le génome humain alors que seulement 2 E1 et environ 40 E2 ont été identifiées à ce jour.
Cependant, l’adressage du substrat poly-ubiquitinylé ne conduit pas nécessairement à la mort de la protéine car il existe une compétition cinétique entre vie et mort des protéines (Collins & Goldberg, 2017) (Figure 2). Une dé-ubiquitinylation peut être réalisée par les dé-ubiquitinases Usp14 ou Uch37 assurant ainsi la survie de la protéine. Au contraire, des activations allostériques impliquant les 6 ATPases situées dans la base de la particule régulatrice 19S provoquent l’ouverture du pore situé à la jonction du corps catalytique 20S et de cette particule régulatrice 19S. La protéine est alors dé-ubiquitinylée par la déubiquitinase Rpn11, capturée dans la particule 20S après passage par le pore ouvert où elle subit une dégradation itérative jusqu’au stade de peptides (2 à 22/23 acides aminés) (Reboud-Ravaux, 2021). L’étape de reconnaissance des substrats protéiques par les E3 est donc essentielle pour aboutir à leur dégradation sélective.
Contrôle de l’UPS par des petites molécules exogènes. Le contrôle de l’action dégradative de protéines particulières par l’UPS est de grand intérêt puisque beaucoup de pathologies héritées ou acquises sont liées au fonctionnement anormal d’une protéine donnée. Le domaine très actif des inhibiteurs du protéasome constitutif et de l’immunoprotéasome (Figure 2) a conduit à des molécules utilisées en thérapeutique (cancers liquides) et est porteur de grands espoirs dans les processus anti-inflammatoires, les désordres auto-immuns et le rejet de greffes (Reboud-Ravaux, 2021). Plusieurs autres composés sont en étude préclinique pour l’inhibition des enzymes impliquées dans l’UPS : inhibiteurs de DUB, de E1, de E2 et surtout de nombreuses E3 (Chamberlain & Cathers, 2019 ; Wu et al., 2020). Parmi les E3 ciblées par des inhibiteurs, on trouve cereblon (CRBN), MDM2, les IAP et DCAF15. À côté d’inhibiteurs du site actif de E3, il y a quelques exemples de ciblage de l’interaction entre une E3 et une E2 (Blaquiere et al., 2020 ; Wu et al., 2020) ou une E3 et un substrat ce qui, dans les deux cas, empêche l’ubiquitinylation de ce substrat protéique et donc sa dégradation (Bowen et al., 2017). L’importance des E3 dans la recherche de molécules à visée thérapeutique s’est trouvée renforcée par le développement de la technologie PROTAC puisque les E3 sont les vecteurs utilisés pour provoquer la dégradation ciblée de protéines spécifiques.
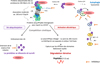 |
Figure 2 La compétition cinétique entre vie et mort des protéines gérée par l’UPS. Les différentes possibilités d’inhibition par de petites molécules exogènes de la dégradation de protéines sont indiquées : inhibition des enzymes E1, E2 et E3 ; inhibition de l’interaction E2-E3 ; inhibition du recrutement du substrat protéique par E3 ; inhibition des différentes activités catalytiques du protéasome ; inhibition des activités dé-ubiquitinase et ATPasique du protéasome 26S ; inhibition de la fixation de la chaîne de poly-ubiquitine par les sous-unités Rpn1, 10 et 13 de la particule régulatrice 19S. La protéine poly-ubiquitinylée liée au protéasome 26S peut être soit dé-ubiquitinylée (elle survit), soit dégradée en peptides à la suite d’une activation allostérique. |
Les molécules PROTAC : d’une curiosité chimique vers une nouvelle catégorie de médicaments
Alors que des petites molécules sont utilisées pour inhiber l’action d’une protéine en bloquant un site actif ou un site essentiel d’interaction, les petites molécules PROTAC cooptent la machinerie cellulaire pour dégrader complètement la protéine cible. Les PROTAC sont des petites molécules hétéro-bifonctionnelles comportant deux ligands : l’un est susceptible de se fixer sur une ubiquitine ligase E3 donnée et l’autre sur la protéine d’intérêt que l’on veut éliminer pour des raisons thérapeutiques (Figures 3A et 3B). Ces deux ligands sont donc liés au sein d’une même molécule par un espaceur covalent de nature variable dont la composition et la longueur peuvent avoir un effet considérable sur l’efficacité de l’édifice PROTAC (Figure 3B). Après formation d’un complexe ternaire avec une E2, la protéine cible est ubiquitinylée et dirigée vers le protéasome 26S qui peut la dégrader. Les ligases E3 ayant été le plus fréquemment utilisées comme cibles pour la production de PROTAC sont présentées dans le Tableau 1. Les Figures 4 et 5 montrent les structures de quelques PROTAC caractéristiques.
En 1999, un premier brevet décrivant le concept de dégradation ciblée d’une protéine a été déposé par la firme Biotech Proteinix, mais il est resté sans suite (Kenten et al., 2000). Il a été suivi deux ans plus tard par les travaux de Craig Crews (Université de Yale) et de Raymond Deshaies (California Institute of Technology). Le recrutement de la E3 SCFβ-TRCP par un peptide a conduit en 2001 à l’induction de l’ubiquitinylation de la méthionine aminopeptidase 2 (MetAP-2) et à sa dégradation par le protéasome 26S (Sakamoto et al., 2001). Ces auteurs ont introduit le terme PROTAC ou PROteolysis Targeting Chimeras pour ce type de molécules induisant la dégradation, terme maintenant préférentiellement utilisé. Cependant, d’autres dénominations peuvent être trouvées dans la littérature pour désigner ces objets : dégradeurs, dégroimides (pour des molécules imides), PROTAP, SNIPER, PDP. La dégradation des récepteurs des œstrogènes (RE) et des androgènes (RA) est réalisée en 2003 (Sakamoto et al., 2003). Un fragment du peptide HIF-1α a permis de recruter la ligase VHL en 2004 (Figure 4) (Min, 2002) alors que le premier PROTAC ciblant MDM2 et basé sur une petite molécule non peptidique est créé en 2008. Cette molécule PROTAC non peptidique peut se lier à la fois au MDM2 (ligase E3) et au RA (Schneekloth et al., 2008). Le nombre de molécules PROTAC nouvellement conçues a ensuite augmenté très rapidement impliquant d’autres E3 et d’autres protéines-cibles à dégrader (Figure 1C) (Petterson & Crews, 2019 ; Wang et al., 2020). On peut citer, de manière non exhaustive, les travaux de Hashimoto et al. pour le premier recrutement de cIAP1 ciblant des protéines liant le rétinol et l’acide rétinoïque (Itoh et al., 2010) et ceux de Ciulli et al. pour les études de relations structure-activité pour des ligands de VHL (Soares et al., 2018 ; Testa et al., 2018). Les potentialités thérapeutiques de cette technologie ont conduit Crews, en 2013, à fonder la société Arvinas. À la même époque, Bradner (Dana-Faber Cancer Institute, puis Novartis) et collègues créent des « dégradeurs » qu’ils appellent « dégroimides » pouvant provoquer la dégradation des protéines de la famille BET (Fischer et al., 2014 ; Gandhi et al., 2014 ; Krönke et al., 2014). Ils ont en effet observé que des molécules imides immunomodulatrices (thalidominide, lenalidomide et pomalidomide dont la structure est présentée sur la Figure 8A) se lient à la ligase E3 cereblon (CRBN) (Lu et al., 2014). Ils créent la société C4 Therapeutics. Ainsi des molécules qui semblaient « amusantes » se sont révélées, contre toute attente, présenter des qualités thérapeutiques. Elles ont généralement une bonne solubilité et une bonne pénétration cellulaire, elles peuvent être utilisées oralement, résister aux processus métaboliques et, dans certains cas, traverser la barrière hématoencéphalique. Les propriétés de ces molécules ne sont en effet pas la somme de celles des différentes parties qui les constituent. La société C4 Therapeutics cherche actuellement à optimiser ce type de molécules, non seulement en amenant à proximité la cible et la ligase, mais aussi en activant l’UPS. Diverses variations de PROTAC ont été développées comme les HaloPROTAC (Buckley et al., 2015) et les CLIPTAC (Astex Pharmaceuticals) qui seront présentés plus loin (Lebraud et al., 2016).
Plusieurs PROTAC sont en cours d’essai clinique. Deux sont pilotés par la Société Arvinas concernant la molécule ARV-110 pour le cancer de la prostate et la molécule ARV-471 pour le cancer du sein (Tableau 2) (Mullard, 2019a, b). Il s’agit de molécules bifonctionnelles dont un bras se fixe sur la protéine-cible (RA pour ARV-110 et RE pour ARV-471) alors que l’autre bras se fixe sur une ubiquitine ligase E3. La molécule ARV-471 (structure non dévoilée) est en phase clinique I chez des femmes présentant un cancer du sein avancé ou métastatique positif ER/négatif HER2 (Flanagan et al., 2018) ; la molécule ARV-110 (structure non dévoilée) l’est pour les cancers résistants de la prostate (Neklesa et al., 2019). À côté des Biotechs (Arvinas, C4 Therapeutics, Kymera Therapeutics), les grands groupes pharmaceutiques investissent massivement dans cette technologie. On peut citer les PROTAC ciblant Bcl6 (Astra Zeneca ; McCoull et al., 2018) et les tyrosine kinases BTK (Pfizer ; Zorba et al., 2018), FAK (Boehringer ; Popow et al., 2019), et IRAK (Glaxo Smith Kline ; Nunes et al., 2019).
 |
Figure 3 Représentation schématique de la dégradation ciblée d’une protéine induite par une molécule PROTAC dans un contexte cellulaire. A. Schématisation de l’ensemble du processus. La molécule PROTAC bifonctionnelle comporte un élément capable de recruter une ligase E3 et un élément recrutant la protéine à dégrader reliés par un espaceur. En se fixant sur ces deux cibles, la molécule PROTAC induit la formation d’un complexe protéine-PROTAC-E2-E3 permettant à la ligase d’ubiquitinyler la protéine d’intérêt sur un résidu lysine de surface. Quand la protéine d’intérêt est suffisamment ubiquitinylée, elle peut être dégradée par le protéasome 26S. À noter, le recyclage du PROTAC en accord avec son action catalytique. B. Schématisation de la taille relative d’une molécule PROTAC par rapport à celle d’une E3 et d’une protéine-cible. |
Exemples de E3 ligases utilisées couramment dans la production de PROTAC. Leurs ligands sont indiqués ainsi le type de protéines qui ont été ciblées.
 |
Figure 4 Exemples de molécules PROTAC avec indication de la nature de la protéine-substrat et de la ligase E3 ciblée. |
 |
Figure 5 Composition des PROTAC et schématisation de leur conception. A. Recrutement par un PROTAC original de différentes protéines-cibles impliquées dans des pathologies via différentes ubiquitine ligases E3. Les protéines sont alors ubiquitinylées et dirigées vers le protéasome 26S qui catalysera leur dégradation. Les molécules PROTAC sont constituées d’un ligand spécifique de la protéine à cibler, d’un espaceur de diverse nature et d’un ligand spécifique d’une E3 donnée. B. Élaboration rationnelle des PROTAC pouvant se lier à différentes E3 (VHL, MDM2, CRBN et ClAP1) tout en utilisant différents ligands de protéines spécifiques via des espaceurs de structure variable. |
PROTAC et colles moléculaires en préclinique et en clinique (adapté de Chamberlain & Hamann, 2019).
Caractéristiques structurales des PROTAC. Leur conception et leur optimisation
La technologie PROTAC repose sur le rationnel suivant : déclencher artificiellement l’induction de la dégradation d’une protéine donnée en amenant à proximité deux protéines (cette protéine et une E3) qui, normalement, n’interagissent pas. Le PROTAC doit donc avoir une affinité adéquate à la fois pour l’E3 choisie et la protéine d’intérêt.
Les ubiquitine-ligases E3. Trois grandes familles de ligases E3 existent : la famille des RING/Ubox, la famille HECT contenant une cystéine dans le site actif et celle dénommée RBR (Zheng & Shabek, 2017 ; Taillandier, 2021). Plusieurs E3 ligases ont été employées pour concevoir des PROTAC telles que la machinerie CRL ou encore de simples RING E3 ligases comme MDM2 et cIAP1 (Tableau 1). De nouveaux PROTAC ont été élaborés lorsqu’on a découvert que les médicaments immunomodulateurs IMiD étaient des ligands de CRBN, un récepteur de substrat de l’E3 ligase Cul4-RING. La spécificité de cette ligase se trouve changée et elle peut dégrader des néo-substrats tels que les facteurs de transcription IKZF1 et IKZF3 (Gandhi et al., 2014 ; Krönke et al., 2014). La Figure 5A illustre la possibilité d’utiliser différentes E3 pour recruter différentes protéines-cibles, et ceci, à chaque fois, en élaborant un PROTAC de structure unique (Sakamoto et al., 2001). Des exemples concrets de combinaison pour créer des PROTAC sont présentés sur la Figure 5B (Cromm & Crews, 2017). À noter que la ligase VHL reconnaît HIF1-α qui présente un résidu proline clé (Figures 4 et 5) : il fait partie d’un motif de reconnaissance spécifique de dégradation (appelé dégron) et est un élément essentiel pour le recrutement de VHL.
Pour l’instant, peu de ligases ont été ciblées parmi les 600 ligases putatives existantes (Zheng & Shabek, 2017). Si seules cinq à six ligases sont validées (Lai & Crews, 2017 ; Wang et al., 2020), des listes beaucoup plus longues existent et plusieurs font l’objet d’études car il convient d’augmenter le recrutement de nouvelles E3 (Fisher & Phillips, 2018 ; Petterson & Crews, 2019 ; Zou et al., 2019 ; Wang et al., 2020 ; Bond & Crews, 2021). En effet, les E3 ont un profil unique en termes d’activité et de distribution dans l’organisme. Leur architecture et leur profil d’expression ont été systématiquement analysés (Schapira et al., 2019). On peut choisir une ligase exprimée de manière ubiquitaire ou, au contraire, une ligase exprimée dans un tissu ou un type de cellule cancéreuse particulier ou même une E3 ayant une localisation subcellulaire particulière. Si une E3 a une expression liée à une pathologie, des PROTAC la ciblant auront en effet une cytotoxicité diminuée puisque la protéine sera épargnée dans les cellules saines. D’importants travaux dans ce domaine portent sur les protéines de la famille MAGE afin d’induire la dégradation spécifique d’une protéine dans une tumeur (Pineda et al., 2015 ; Bond & Crews, 2021).
Les protéines ciblées. Une grande variété de protéines a été ciblée. Elles appartiennent à plusieurs catégories. Les kinases sont très représentées : RIPK2, Fak, BCR-ABL, BTK, HER2, PI3K, c-Met, CDK9, TBK1, ALK, Akt, ERK1/2, FLT3. Les récepteurs nucléaires des androgènes (RA) et des œstrogènes (RE) ont conduit aux deux PROTAC actuellement en étude clinique (Tableau 2). Les protéines BET impliquées dans la régulation de la transcription BDR4 et BDR9 sont aussi des cibles de choix. Différentes autres protéines comme des enzymes (HDAC6, MetAp-2), des protéines liées au système nerveux (Tau, α-synucléine, Huntingtine, Sirt2), des récepteurs (EGFR), des protéines régulatrices (Bcl-2) ou encore SMAD3 par exemple sont également des cibles potentielles pour des applications de la technologie PROTAC (Petterson & Crews, 2019). Des résultats positifs ont déjà été obtenus avec des protéines connues pour être difficiles à cibler en vue de l’obtention de médicaments. Parmi elles, on trouve FRS2α (protéine d’échafaudage), les complexes protéiques BAF/PBAF (SMARCA2/4) (Farnaby et al., 2019 ; Kargbo, 2020), des facteurs de transcription provoquant des changements dans l’expression des gènes (STAT3), des protéines de la famille Ras (KRAS G12C) (Bond & Crews, 2021). Ces résultats devraient aider au ciblage futur de cMyc et d’autres mutants de Ras.
Les espaceurs. La nature de l’espaceur est importante car elle joue sur l’activité biologique et les propriétés physico-chimiques de la molécule de PROTAC. On en trouve de différents types : linéaires ou cycliques, flexibles ou relativement rigides (Bondeson et al., 2018 ; Wang et al., 2020) (Figures 4 et 5). Leur optimisation est extrêmement importante car elle permet d’améliorer la perméabilité cellulaire du PROTAC, sa distribution tissulaire et son métabolisme. Des espaceurs peptidiques ont été initialement utilisés. Actuellement, des PEG de longueur variable et des alcanes saturés ou insaturés sont communément utilisés. La présence d’oxygène dans l’espaceur peut stabiliser l’orientation d’une protéine alors qu’un groupe pipéridine peut augmenter la solubilité du PROTAC. Des espaceurs comme les triazoles sont formés à l’intérieur même de la cellule par chimie Clic (cf. § Assemblage intracellulaire). La longueur de l’espaceur est elle aussi importante : trop courte, les deux protéines ne peuvent se lier simultanément au PROTAC par suite d’une gêne stérique ; trop longue, l’interaction entre les deux protéines n’est plus possible.
Données structurales et aide à la conception. La structure de 10 complexes ternaires différents est connue (Gadd et al., 2017 ; Nowak et al., 2018 ; Farnaby et al., 2019 ; Roy et al., 2019 ; Testa et al., 2020 ; Schiemer et al., 2021). Il est encore à l’heure actuelle difficile de définir des principes généraux permettant d’obtenir des complexes ternaires efficaces (Konstantinidou et al., 2019 ; Petterson & Crews, 2019). Pourtant, les PROTAC peuvent être considérés comme des activateurs de dégradation programmables car concevables pour n’importe quelle protéine cible. Indispensables pour son ubiquitinylation, ils interviennent dans la formation d’un complexe ternaire de façon catalytique. Les études cristallographiques, biophysiques et mécanistiques décrites dans le paragraphe suivant ont montré que l’analyse seule de l’affinité des PROTAC au sein des complexes binaires était insuffisante pour guider l’élaboration de PROTAC efficaces ; la coopérativité dans le complexe ternaire est importante mais peut différer selon la nature de l’E3 et de la protéine cible. La conception rationnelle utilisant la modélisation moléculaire conduit à des PROTAC efficaces (Nowak et al., 2018). Alors que les ligands classiques doivent présenter une haute affinité pour un site dédié, le PROTAC a la possibilité de se fixer n’importe où sur la cible d’où les possibilités du criblage biophysique permettant d’augmenter la probabilité de trouver des ligands compatibles. Des banques de ligands ont été approuvées par la FDA et des banques PROTAC sont utilisées pour augmenter l’identification de molécules efficaces (Bond & Crews, 2021). Des plateformes dédiées se développent permettant de réaliser de manière itérative conception, synthèse, test et analyses (Churcher, 2018 ; Nishiguchi et al., 2021). Les modélisations mathématiques et l’élaboration computationnelle de PROTAC ont conduit à la mise au point de programmes (MOE et Rosetta) (Drummond et al., 2020) donnant des résultats très encourageants ainsi qu’à celui du nouveau programme PRosettaC (Zaidman et al., 2020).
Mécanisme d’action des PROTAC
L’induction de la dégradation d’une protéine par une molécule PROTAC dépend de la formation du complexe ternaire E3-PROTAC-protéine cible puisqu’il permet l’ubiquitinylation de cette protéine et sa dégradation par le protéasome (An & Fu, 2018 ; Petterson & Crews, 2019). Les modèles mathématiques applicables à la formation de complexes ternaires prédisent une variation de leur concentration en fonction de la concentration de PROTAC (Figure 6). De trop fortes concentrations de PROTAC conduisent à des complexes binaires inopérants pour une dégradation induite par le protéasome. On parle alors d’effet « hameçon » observé expérimentalement dans plusieurs cas (Olson et al., 2018 ; Schiedel et al., 2018). Par ailleurs le complexe ternaire formé doit être stable pour permettre l’ubiquitinylation de la protéine cible. Cette stabilité est favorisée par des interactions de coopérativité positive entre l’E3 et la protéine (α > 1) alors qu’une coopération négative due à de l’encombrement stérique ou à des répulsions de charges (α < 1) est défavorable (Figure 6). La coopérativité positive diminue l’effet « hameçon » (Roy et al., 2017). La dégradation induite par un PROTAC présentant une grande promiscuité avec une cinquantaine de kinases a montré que l’efficacité dégradative observée dans un certain nombre de cas était corrélée à la formation de complexes ternaires stables et non à l’affinité du PROTAC pour la cible (Bondeson et al., 2018). Une faible affinité kinase-PROTAC (Kd = 11 μM pour kinase p38α/ PROTAC) peut être compensée par la formation de complexes ternaires stables induisant fortement la dégradation (DC50 = 210 nM, Dmax = 91 %). La première structure cristallographique d’un complexe ternaire a été obtenue par Ciulli et al. en 2017 (Gadd et al., 2017). Le PROTAC MZ1 est lié au bromodomaine BRD4BD2 et à la ligase VHL (MZ1, Figure 4). Lorsqu’elle est engagée dans les cavités de liaison à VHL et à BDR4 au sein du complexe ternaire, la molécule MZ1 se replie sur elle-même ce qui permet une interaction étendue entre les deux protéines (Gadd et al., 2017). La coopérativité positive a été démontrée par diverses techniques biophysiques. Cependant, la coopérativité peut avoir moins d’importance : c’est le cas des PROTAC ciblant les protéines BTK et BDR4 qui, toutes deux, recrutent la ligase CBRN (Nowak et al., 2018). Cette étude a aussi montré l’importance de la nature du ligand qui peut ou non favoriser la formation du complexe ternaire.
 |
Figure 6 Formation en fonction de la concentration de PROTAC d’un complexe ternaire E3-PROTAC-protéine ou de complexes binaires (effet « hameçon »). Une coopération positive entre protéine et E3 (α > 1) sera favorable à l’ubiquitinylation de la protéine contrairement à une coopération négative (α < 1). |
Propriétés ADME des PROTAC et stratégies d’amélioration de leur biodisponibilité
La structure inhabituelle des PROTAC a soulevé beaucoup de questions au sein de la communauté des concepteurs de médicaments (Churcher, 2018). Elle nécessite des évaluations précises en ce qui concerne la solubilité et la stabilité des PROTAC étudiés, leur perméabilité cellulaire, leur faculté à former des complexes binaires et des complexes ternaires avec ou non coopérativité, la détection de l’ubiquitinylation de la protéine-cible (spectrométrie de masse, western blots par exemple) et la dégradation effective de la cible (DC50). Une étude in cellulo en temps réel a été réalisée permettant de suivre la pénétration cellulaire, la formation du complexe ternaire, l’ubiquitinylation et la dégradation de la protéine-cible (Riching et al., 2018). On répond maintenant par l’affirmative à un certain nombre des questionnements de départ comme la pénétration cellulaire (malgré une taille de 700–1000 Da), la stabilité métabolique, la sélectivité de dégradation. Cependant si la destruction complète de la protéine est avantageuse dans certains cas, en particulier pour contrer des effets prolifératifs, dans d’autres cas il faut veiller à ce que l’élimination de la protéine n’entraîne pas la disparition de fonctions essentielles (Saenz et al., 2017).
Le répertoire des protéines-cibles comme celui des E3 continuant à augmenter, la caractérisation préclinique in vitro des PROTAC (ADME et propriétés physico-chimiques) et leur évaluation in vivo au niveau de la pharmacocinétique (PC) et de la pharmacodynamique (PD) sont essentielles. Il convient aussi d’établir une relation quantitative entre la disparition de la protéine-cible et la fonction biologique in vivo. La dégradation de protéines par des PROTAC a été observée dès 2015 par deux groupes indépendants qui ciblaient ERRα et BET en utilisant respectivement les E3 VHL et CUL4CRBN (réduction > 40–50 % de la concentration de la protéine ciblée) (Bondeson et al., 2015 ; Winter et al., 2015). L’étude in vivo de plusieurs PROTAC et la détermination de leurs propriétés PC/PD ont confirmé leur efficacité sur des tumeurs pouvant atteindre 80 % à des doses convenables (Watt et al., 2019). Bien que l’on ne dispose actuellement que de peu d’exemples in vivo (en dehors de la réduction du volume de tumeurs et de l’augmentation de la survie de souris), ces études confirment le potentiel des PROTAC pour offrir une alternative aux inhibitions conventionnelles. L’obtention, dans les études précliniques, de données PC/PD est essentielle et a été recherchée (Tutland et al., 2014). Les particularités de la technologie PROTAC font l’objet d’études de modélisation afin d’aider la recherche translationnelle et d’obtenir une meilleure prédiction des doses cliniques (Wang et al., 2020). Des molécules efficaces par prise orale sont recherchées (Nowak & Jones, 2021).
Assemblage intracellulaire des PROTAC
Bien qu’étant des petites molécules, les PROTAC ont une taille relativement élevée (environ 700–1000 Da). Elles n’obéissent donc pas à la « règle des cinq » de Lipinski, en particulier celle d’une taille inférieure à 500 Da (Lipinski et al., 1997) et autres règles d’intérêt (Walters, 2012). Cette règle, bien que non strictement indispensable, est considérée comme conduisant à des propriétés biologiques et pharmacologiques favorisant le développement de médicaments administrables par voie orale. Une alternative intéressante est d’utiliser la chimie pour faire synthétiser le PROTAC à l’intérieur même de la cellule en fournissant à celle-ci les ligands modifiés et donc nécessairement de taille inférieure au PROTAC. Des combinaisons bi-orthogonales de molécules utilisant la chimie Clic permettent de synthétiser à l’intérieur de la cellule une molécule PROTAC alors dite CLIPTAC (Figure 7). La formation intracellulaire d’un PROTAC a notamment été obtenue par la réaction Clic entre un ligand de la sirtuine 2 porteur d’une fonction alcyne et un dérivé azide de la thalidomide (Figure 8A) (Schiedel et al., 2018). Les protéines BDR4 et ERK1/2, cibles oncologiques clés, ont été dégradées avec succès en traitant des cellules HeLa en culture de manière séquentielle par le ligand modifié de la protéine-cible, puis par le ligand capable de recruter la ligase E3 (ici CRBN) (Figure 8B) (Lebraud et al., 2016).
 |
Figure 7 Stratégie générale de synthèse intracellulaire de PROTAC en utilisant la chimie Clic par cycloaddition alcyne-azide avec catalyse par Cu(I) (A) ou cycloaddition de Diels-Alder entre une tétrazine et un alcène contraint (B). Ces réactions sont très sélectives et n’interfèrent pas avec les processus biochimiques. Les constantes de vitesse des réactions chimiques sont de l’ordre de 102 M−1.s−1(A) et de 104 M−1.s−1(B). Les chimères ainsi synthétisées à l’intérieur de la cellule sont appelées CLIPTAC et ont été utilisées pour les E3 VHL (A) et CRBN (B). Les ligands modifiés devant pénétrer dans la cellule ont un poids moléculaire très inférieur à celui du PROTAC. |
 |
Figure 8 Comparaison entre les stratégies de dégradation induite par les PROTAC et par marquage hydrophobe (colle moléculaire) basé ou non sur HaloTag. A. Schématisation de l’interaction entre une protéine-cible et une E3 (CRBN) médiée par une colle moléculaire (a) ou un PROTAC (b). Le poids moléculaire de la colle moléculaire est inférieur à celui du PROTAC. B. Dégradation d’une protéine cible induite par une molécule HyT conduisant à un repliement incorrect. La molécule HyT provoque la déstabilisation de la conformation de la protéine avec complexation à HSP70 (modèle 1) ou une complexation directe avec HSP70 (modèle 2) conduisant dans les deux cas à une dégradation par le protéasome 26S. Le fonctionnement physiologique pour une protéine mal conformée est indiqué pour comparaison. C. Élaboration d’une molécule HyT ciblant la protéine Tau. Un motif hydrophobe adamantyle ou Boc3Arg est lié par covalence à un peptide reconnaissant spécifiquement Tau, le motif poly-arginine favorisant la pénétration cellulaire. La structure d’un PROTAC dégradant la protéine Tau est indiquée pour comparaison. D. Marquage hydrophobe sur une protéine de fusion HaloTag. La protéine de fusion « protéine d’intérêt-HaloTag » est reconnue par une molécule bifonctionnelle. L’état partiel de dénaturation qu’elle induit provoque la dégradation par le protéasome. |
Les atouts de la stratégie PROTAC
Mécanisme catalytique à des concentrations subnanomolaires. Lors de son action, le PROTAC n’est pas dégradé mais recyclé (Figure 2). Il peut donc à nouveau ubiquitinyler et dégrader de multiples autres protéines-cibles. Ainsi, il opère à des concentrations sub-stoechiométriques (Bondeson et al., 2015). Il peut réduire jusqu’à 90 % le niveau intracellulaire d’une protéine et ceci, à des concentrations nanomolaires. Du fait de leur recyclage, les PROTAC peuvent donc avoir des effets de longue durée.
Contourner la résistance observée dans les cancers. Les inhibiteurs de kinases font partie de l’arsenal chimiothérapique classique du traitement de cancers (Wu et al., 2015). Cependant, des résistances à ce type de traitement sont fréquemment observées. L’avantage des PROTAC est de permettre la dégradation de toutes les protéines-cibles de même nature. Ainsi, la résistance à l’enzatulamide peut être surmontée avec la molécule ARCC-4 ciblant le récepteur androgénique (Salami et al., 2018). Il en est de même avec la molécule L18I ciblant BTK dans les lymphomes non-Hodgkin pour la mutation C481S (Sun Y. et al., 2019).
Élimination à la fois de l’activité enzymatique et des fonctions non enzymatiques de la protéine ciblée. Un autre avantage des molécules PROTAC est d’éliminer non seulement l’activité enzymatique d’une protéine-cible mais aussi d’affecter ses fonctions non enzymatiques puisque la protéine est totalement dégradée. Ceci a été observé pour la kinase FAK en ce qui concerne ses rôles non enzymatiques (Cromm et al., 2018). Un PROTAC sélectif de FAK (IC50 = 6,5 nM ; DC50 = 3 nM ; Dmax = 99 %), basé sur la molécule defactinib utilisée en clinique, altère la fonction de transduction du signal de FAK dépendante de sa fonction kinase mais aussi la transduction du signal indépendante de cette activité enzymatique (Cromm et al., 2018).
Dégradation des protéines ne pouvant être ciblées par les techniques traditionnelles. LA FDA a approuvé des médicaments pour environ 400 protéines alors qu’environ 3000 sont associées à des pathologies. Beaucoup d’entre elles n’ont pas de site actif ou de site de reconnaissance de ligands Les techniques traditionnelles pour la découverte de médicaments ne ciblent que 20 à 25 % des protéines qui sont notamment des enzymes, des récepteurs nucléaires d’hormones, des récepteurs couplés aux protéines G, des canaux ioniques. Les autres sont considérées comme non « ciblables » (Kim et al., 2014). La technologie PROTAC permet donc d’élargir considérablement le nombre de cibles thérapeutiques. Ainsi, une molécule PROTAC a démontré une activité biologique importante in vitro et in vivo, due à son action dégradative de STAT3 avec régression tumorale complète (Bai et al., 2019).
Stratégie d’extinction chimique rapide et réversible. Contrairement à ce qui est observé avec les technologies basées sur les acides nucléiques comme CRISPR-Cas9 par exemple, les PROTAC, au lieu d’agir au niveau du génome, agissent directement sur les protéines en les dégradant. Ceci est un avantage dans l’étude de protéines embryonnaires létales par exemple. Les PROTAC permettent d’autre part d’obtenir la disparition de la protéine-cible de manière contrôlée dans le temps. La suspension du traitement PROTAC permet de récupérer une protéine fonctionnelle. Au contraire, les techniques génétiques d’extinction ont un mode d’action irréversible, de longue durée et coûteux. La stratégie PROTAC apparaît donc comme un outil efficace qui peut être complémentaire de ces techniques (Sun X. et al., 2019).
Autres stratégies de dégradation induite des protéines
La stratégie PROTAC commencée il y a 20 ans connaît actuellement une accélération considérable avec une complexification des stratégies possibles. Parmi celles-ci, on peut citer la formation intracellulaire de PROTAC par la chimie Clic (voir plus haut), les colles moléculaires, le marquage hydrophobe, les haloPROTAC, les homo-PROTAC et les ARN-PROTAC.
Colles moléculaires. Les PROTAC favorisent la dégradation d’une protéine spécifique via les E3 en permettant la proximité entre cette protéine et une E3 particulière pendant un temps de résidence suffisant (Figure 8A,b). Des molécules distinctes des PROTAC ne présentant pas deux ligands séparés par un espaceur peuvent cependant structurer une telle interaction. Un processus de ce type est observé avec les hormones de plantes (auxine et jasmonate) qui structurent les complexes ligase-substrat d’où le terme de « colle moléculaire » donné à de telles molécules (Tan et al., 2007 ; Sheard et al., 2010). Bien que possédant une affinité faible pour leurs ligases respectives et les substrats protéiques isolés, l’affinité des colles moléculaires au sein du complexe ternaire est suffisante pour permettre l’ubiquitinylation des substrats et la dégradation par le protéasome (Figure 8A,a). En 2012, la dégradation de la glutathion-S-transférase (GST) induite par son inhibiteur covalent, l’acide étacrynique, lié par covalence à Boc3Arg a été réalisée (Long et al., 2012). De même, la DHFR a été dégradée avec des concentrations micromolaires de son inhibiteur non covalent, le triméthoprime, couplé à Boc3Arg (Long et al., 2012). Les ligands porteurs du groupe hydrophobe Boc3Arg induisent aussi la dégradation par le protéasome 20S (Shi et al., 2016). Plusieurs colles moléculaires ont été approuvées en tant que médicaments, d’autres sont en phase clinique I ou II pour le traitement de myélome, de lymphome et du lupus (Tableau 2) (Chamberlain & Hamann, 2019). L’exploitation de nouvelles données structurales et l’utilisation d’espaceurs minimaux amèneront peut-être à estomper les différences entre PROTAC et colles moléculaires (Chamberlain, 2018).
Dégradation de protéines par induction de repliement incorrect. L’objectif de cette stratégie est de déstabiliser la protéine-cible par marquage, ce qui revient à mimer un mauvais repliement (Neklesa & Crews, 2012). Elle est alors dirigée vers le protéasome puisque celui-ci constitue la machinerie endogène responsable du contrôle de la qualité des protéines.
Le marquage hydrophobe (HyT). L’étiquette HyT est un motif hydrophobe (adamantane ou Boc3Arg par exemple) lié à un ligand de la protéine-cible (Figure 8B). Dans un premier type de mécanisme, HyT déstabilise la protéine ce qui permet de recruter une protéine chaperonne conduisant à la dégradation par le protéasome 26S (Figure 8B, modèle 1). Dans un autre mécanisme, la molécule HyT est directement reconnue par la chaperonne conduisant à la dégradation protéosomale (Figure 8B, modèle 2) (Wang et al., 2020). Les chaperonnes de la famille des HSP70 reconnaissent en effet des régions hydrophobes de protéines qui se trouvent exposées lorsque celles-ci ont une mauvaise conformation (Figure 8B, fonctionnement physiologique). À noter que le protéasome 20S peut aussi dégrader une protéine combinée à une molécule HyT (Wang et al., 2020). Des molécules HyT constituées d’un motif hydrophobe et d’un peptide se liant spécifiquement à une protéine-cible (Figure 8C) peuvent induire la dégradation d’agrégats de protéines (Aβ et Tau) (Suh et al., 2007). Ainsi, une dégradation sélective de la protéine Tau dépendant à la fois de la concentration et du temps a été observée dans le cerveau d’un modèle murin de la maladie d’Alzheimer. Des traçages par tomographie par émission de positons (TEP) ont pu en être réalisés (Silva et al., 2019).
Le marquage hydrophobe (HyT) basé sur HaloTag. HaloTag est une déhalogénase bactérienne (297 acides aminés) modifiée qui se lie par covalence à un ligand synthétique chloroalcane. La protéine HaloTag liée à la protéine d’intérêt peut être recrutée par une molécule bifonctionnelle contenant une chaîne chloroalkyle et un ligand hydrophobe qui se lie à la protéine. Ceci provoque un état de dénaturation partielle de la protéine déclenchant la machinerie de contrôle par le protéasome (Figure 8D) (Neklesa et al., 2011)
Il est à noter qu’un marquage HaloTag peut provoquer la dégradation d’une protéine de fusion par le protéasome en présence d’une molécule bifonctionnelle contenant une chaîne chloroalkyle (se liant par covalence à la protéine de fusion HaloTag) et un ligand d’une E3. Ces deux motifs amènent la protéine de fusion HaloTag à proximité de la ligase E3 permettant sa dégradation (Buckley et al., 2015 ; Tomoshige et al., 2016).
Les homo-PROTAC. On crée un PROTAC comportant deux ligands d’une E3 ce qui revient à créer un dimère ciblant cette E3 et à déclencher alors son élimination suicide à l’intérieur de la cellule (Figure 9A). Ont été ainsi réalisées l’auto-dégradation de VHL (Maniaci et al., 2017) et celle de MDM2 (He et al., 2020). Dans ce dernier cas, une activité anti-tumorale efficace a été démontrée chez la souris atteinte d’un cancer pulmonaire non à petites cellules.
 |
Figure 9 Homo-PROTAC et RIBOTAC. A. Structure d’un homo-PROTAC ciblant la E3 ligase MDM2 (1) et d’un homo-PROTAC ciblant la E3 ligase VHL (2). Ces Homo-PROTAC ont été conçus pour provoquer l’auto-dégradation par le protéasome de chacune de ces ligases. B. Schématisation du mode d’action d’un RIBOTAC. |
Extension de la dégradation ciblée à d’autres constituants cellulaires
Des petits mimes d’ARN, les ARN-PROTAC, sont susceptibles de se lier à l’ARN et à une E3 et ainsi d’induire la dégradation de protéines se liant à l’ARN (Ghidini et al., 2021). Par ailleurs, ont été créées des molécules hétéro-bifonctionnelles formées d’un ligand pouvant se lier à un ARN attaché via un espaceur à un autre ligand pouvant se lier et activer une ARNase (Costales et al., 2019). Ces chimères appelées RIBOTAC permettent de dégrader l’ARN lui-même (Figure 9B). La dégradation d’un ARN par une ribonucléase ainsi recrutée par une petite molécule a permis d’inhiber à un niveau sub-stoechiométrique la métastase d’un cancer du poumon dans un modèle murin (Costales et al., 2019, 2020). De même, la dégradation de l’ARN viral du SARS-CoV-2 a été obtenue à l’aide d’un RIBOTAC (Haniff et al., 2020). On estime qu’il y a 100 fois plus d’ARN que de protéines-cibles. Cependant le ciblage d’ARN est rare et difficile (Costales et al., 2019).
Des chimères LYTAC ciblant le lysosome ont permis d’induire la dégradation de protéines extracellulaires (Banik et al., 2020 ; Alabi & Crews, 2021). En détournant le système d’autophagie, des chimères MADTAC acquièrent la possibilité de détruire des agrégats de protéines et des organelles entières (Takahashi et al., 2019 ; Ding et al., 2020 ; Alabi & Crews, 2021).
Stratégie de stabilisation des protéines
Tout à fait récemment, des molécules hétéro-bifonctionnelles ont été créées, les DUBTAC, pour stabiliser une protéine. Dans certaines pathologies, on ne recherche pas à éliminer une protéine mais au contraire à la stabiliser. On a ainsi pu stabiliser la molécule DF508-CFTR dans des cellules humaines de fibrose cystique (Henning et al., 2021).
Conclusion
La cellule eucaryote contient de 109 à 1010 protéines impliquées dans de vastes réseaux au sein desquels elles doivent intervenir de manière excessivement précise dans l’espace et dans le temps. L’UPS y joue un rôle central et sa perturbation peut contribuer de manière directe à la survenue et à la progression de nombreuses pathologies (cancers, maladies neurodégénératives, syndrome métabolique, désordres inflammatoires, auto-immunité, dystrophies musculaires, maladies infectieuses). Les composants du système UPS constituent donc, de manière classique, des cibles thérapeutiques mais l’UPS peut aussi être détourné, dans un premier temps, de sa fonction première de protéolyse en vue d’un bénéfice thérapeutique. À ce titre, la technologie PROTAC s’appuie sur une stratégie nouvelle et puissante. Elle a l’originalité d’agir selon un mécanisme de type catalytique avec recyclage de l’agent thérapeutique et ceci à un niveau sub-stoechiométrique. L’action sur la protéine-cible est sélective conduisant à son extinction rapide et réversible, applicable aussi bien in cellulo qu’in vivo. Elle présente deux autres caractéristiques majeures : (a) elle permet d’éliminer une énorme variété de protéines « non ciblables » par les méthodes habituelles utilisées dans le développement de médicaments car elle ne nécessite pas la présence d’un site actif ou d’un site de reconnaissance dédié à un ligand physiologique ; (b) elle utilise les propriétés de dégradation uniques de l’UPS, système ubiquitaire largement conservé chez toutes les espèces. Malgré quelques faiblesses, cette technologie présente donc de grandes forces (Luh et al., 2020).
À l’entrée de la troisième décennie pour cette technologie, on note une accélération considérable et une complexification avec des développements remarquables comme la production d’homo-PROTAC, le marquage hydrophobe, la technologie des HaloPROTAC, les ARN-PROTAC, les RIBOTAC. Au moins 15 PROTAC et colles moléculaires seront en études cliniques à la fin 2021 (Mullard, 2021). On s’oriente donc à l’avenir vers l’élimination de protéines dites difficiles à cibler dans une optique thérapeutique. Le recrutement de nouvelles ligases non encore utilisées sera en pleine expansion. L’élaboration de nouvelles structures de type PROTAC s’appuiera fortement sur les analyses des interfaces non naturelles entre protéines en utilisant des données structurales et de nouvelles méthodes informatiques comme boîtes à outils.
Abréviations
cABL : Cellular form of the Abelson leukemia virus tyrosine kinase
ADME : Absorption, distribution, métabolisme et excrétion (données de pharmacocinétique)
ARNsi : ARN interférent
Bcl-2 : B-cell lymphoma 2
Bcl-6 : B-cell lymphoma 6
BET : Bromodomain and extra-terminal motif (in BDR proteins)
Boc3-Arg : Tert-butyl carbamate-protected arginine
BRD4 : Bromodomain containing protein 4 (BET family member)
BTK : Bruton’s tyrosine kinase
CDC4 : Cell division control protein 4
CFTR : Cystic fibrosis transmembrane conductance regulator
cIAP1 : Cellular inhibitor of apoptosis protein 1
CLIPTAC : In-cell click-formed-targeting chimera
CRBN : Cereblon
CRISPR-Cas9 : Clustered regularly interspaced short palindromic repeats associated protein 9
CRL : Cullin-Ring E3 ubiquitin ligase
DC50 : Concentration de PROTAC pour laquelle une dégradation égale à 1/2Dmax est atteinte
DCAF15 : DDB1 and CUL4 associated factor 15
DCP : Dégradation ciblée d’une protéine
Dmax : Concentration de PROTAC conduisant à la dégradation maximum d’une protéine
DUBTAC : Deubiquitinase-targeting chimera
E1 : Ubiquitin-activating enzyme
E2 : Ubiquitin-conjugating enzyme
E3 : Ubiquitin ligase
EGFR : Epithelial growth factor receptor
ERRα : Estrogen-related receptor α
FAK : Focal adhesion kinase
FDA : Food and Drug Administration
HDAC6 : Histone deacetylase 6
HECT : Homologous to E6-AP carboxyl terminus
HER2 : Human epidermal growth factor receptor 2
HIF-1α : Hypoxia-inducible factor 1 subunit alpha
HSP : Heat shock protein
IAP : Inhibitors of apoptosis proteins
cIAP1 : Cellular inhibitor of apoptosis protein 1
IC50, IC90-95 : Concentration d’inhibiteur réduisant de moitié (50 %) ou supprimant quasi complètement (90–95 %) la fonction de la protéine ciblée
IKZF1 (ou 3) : IKAROS family zinc finger 1 (or 3)
IMiD : Médicaments immunomodulateurs
IRAK4 : Interleukin-1 receptor-associated kinase 4
LYTAC : Lysosome targeting chimera
MADTAC : Macroautophagy degradation targeting chimera
MAGE : Melanoma associated antigen
MDM2 : Mouse double minute two homolog
MEIS2 : Meis homeobox 2
MOE : Molecular operating environment
cMyc : MYc proto-oncogene, bHLH transcription factor
PC : Pharmacocinétique
PD : Pharmacodynamique
PDP : Protein degradation probe
PEG : Polyéthylène glycol
PROTAC : PROteolysis TArgeting Chimera
PROTAP : PROteolysis TArgeting Peptide
RA : Récepteur d’androgène
RAS : Rat sarcoma proteins
RBR : Ring between ring
RE : Récepteur d’œstrogène
RING : Really interesting new genes
RIPK2 : Receptor-interacting serine/threonine kinase 2
RTK : Tyrosine kinase receptor
SCF : Skp 1-cullin-F-box
SCFβ-TRCP : Mammalian SCF complex recognizing a destruction motif in IkBα
Sirt2 : Sirtuine 2
SMAC : Second mitochondrion-derived activator
SMAD3 : Mothers against decapentaplegic homolog 3
SMARCA2 : WI/SNF related, matrix associated, actin dependent regulator ofc, Subfamily A, member 2
SNIPER : Specific and non-genetic IAP-dependent protein eraser
STAT3 : Signal transducer and activator of transcription 3
Sub-stoechiométrique : Effet plus grand d’une molécule que le rapport 1:1 obtenu avec un agent stoechiométrique
SUP : Système ubiquitine protéasome
VHL : Protéine von Hippel–Lindau
Remerciements
Je remercie Madame Sophie Gournet (Plateforme Illustration & Graphisme ; IBPS, Sorbonne Université) qui a réalisé la représentation graphique de la molécule de protéasome 26S sur la Figure 2.
Références
- Adjei, A.A. (2006). What is the right dose? The elusive optimal biologic dose in phase I clinical trials. J Clin Oncol, 24, 4054-4055. [PubMed] [Google Scholar]
- Alabi, S.B., Crews, C.M. (2021). Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. J Biol Chem, 296, 100647. [PubMed] [Google Scholar]
- An, S., Fu, L. (2018). Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. EBioMedecine, 36, 553-562. [Google Scholar]
- Bai, L., Zhou, H., Xu, R., Zhao, Y., Chinnaswamy, K., McEachern, D., Chen, J., Yang, C.Y., Liu, Z., Wang, M., Liu, L., Jiang, H., Wen, B., Kumar, P., Meagher, J.L., Sun, D., Stuckey, J.A., Wang, S.A. (2019). Potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell, 36, 498-511.e1. [PubMed] [Google Scholar]
- Banik, S.M., Pedram, K., Wisnovsky, S., Ahn, G., Riley, N.M., Bertozzi C.R., (2020). Lysosome-targeting chimeras for degradation of extracellular proteins. Nature, 584, 291-297. [PubMed] [Google Scholar]
- Blaquiere, N., Villemure, E., Staben, S.T. (2020). Medicinal chemistry of inhibiting RING-type E3 ubiquitin ligases. J Med Chem, 63, 7957-7985. [PubMed] [Google Scholar]
- Bond, M.J., Crews, C.M. (2021). Proteolysis targeting chimeras (PROTACs) come of age: entering the third decade of targeted protein degradation. RCS Chem Biol. DOI: 10.1039/D1CB00011J. [Google Scholar]
- Bondeson, D.P., Mares, A., Smith, I.E.D., Ko, E., Campos, S., Miah, A.H., Mulholland, K.E., Routly, N., Buckley, D.L., Gustafson, J.L., Zinn, N., Grandi, P., Shimamura, S., Bergamini, G., Faelth-Savitski, M., Bantscheff, M., Cox, C., Gordon, D.A., Willard, R.R., Flanagan, J.F., Casillas, L.N., Votta, B.J., Den Besten, W., Famm, K., Kruidenier, L., Carter, P.S., Harling, J.D., Churcher, I., Crews, C.M. (2015). Catalyticin vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol, 11, 611-617. [PubMed] [Google Scholar]
- Bondeson, D.P., Smith, B.E., Burslem, G.M., Buhimschi, A.D., Hines, J., Jaime-Figueroa, S., Wang, J., Hamman, B.D., Ishchenko, A., Crews, C.M. (2018). Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem Biol, 25(1), 78-87. [PubMed] [Google Scholar]
- Bowen, T.S., Adams, V., Werner, S., Fischer, T., Vinke, P., Brogger, M.N., Mangner, N., Linke, A., Sehr, P., Lewis, J., Labeit D., Gasch, A., Labeit, S. (2017). Small-molecule inhibition of MuRF1 attenuates skeletal muscle atrophy and dysfunction in cardiac cachexia. J Cachexia Sarcopenia Muscle, 8, 939-953. [PubMed] [Google Scholar]
- Buckley, D.L., Raina, K., Darricarrere, N., Hines, J., Gustafson, J.L., Smith, I.E., Miah, A.H., Harling, J.D., Crews, C.M. (2015). HaloPROTACS: use of small molecule PROTACs to induce degradation of HaloTag fusion proteins. ACS Chem Biol, 10, 1831-1837. [PubMed] [Google Scholar]
- Burslem G.M., Crews, C.M. (2020). Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell, 181, 102-114. [PubMed] [Google Scholar]
- Burslem, G.M., Smith, B.E., Lai, A.C., Jaime-Figueroa, S., McQuaid, D.C., Bondeson, D.P., Toure, M., Dong, H., Qian, Y., Wang, J., Crew, A.P., Hines, J., Crews, C.M. (2018). The advantages of targeted protein degradation over inhibition: an RTK case study. Cell Chem Biol, 25(1), 67-77.e63. [PubMed] [Google Scholar]
- Chamberlain P.P. (2018). Linkers for protein degradation. Nature Chem Biol, 14, 638-641. [Google Scholar]
- Chamberlain, P.P., Cathers, B.E. (2019). Cereblon modulators: low molecular weight inducers of protein degradation. Drug Discov Today, 31, 29-34. [Google Scholar]
- Chamberlain, P.P., Hamann, L.G. (2019) Development of targeted protein degradation therapeutics. Nat Chem Biol, 15, 937-944. [PubMed] [Google Scholar]
- Churcher, I. (2018). PROTAC-induced protein degradation in drug discovery: breaking the rules or just making new ones? J Med Chem, 61, 444-452. [PubMed] [Google Scholar]
- Collins, G.A., Goldberg, A.L. (2017). The logic of the 26S proteasome. Cell, 169, 792-806. [CrossRef] [PubMed] [Google Scholar]
- Conde, J., Artzi, N. (2015) Are RNAi and miRNA therapeutics truly dead? Trends Biotech, 33, 141-144. [Google Scholar]
- Cong, L., Ran, F.A., Cox, D., Lin, S., Baretto, R., Habib, N., Hsu, P.D., Wu, X., Jiang, W., Marraffini, L.A., Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science, 339, 819-823. [CrossRef] [PubMed] [Google Scholar]
- Costales, M.G., Suresh, B., Vishnu, K., Disney, M.D. (2019). Small-molecule targeted recruitment of a nuclease to cleave an oncogenic RNA in a mouse model of metastatic cancer. Cell Chem Biol, 26, 1180-1186. [PubMed] [Google Scholar]
- Costales, M.G., Aikawa, H., Li, Y., Childs-Disney, J.L., Abegg, D., Hoch, D.G., Velagapudi, S.P., Nakai, Y., Khan, T., Wang, K.W., Yildirim, I., Adibekian, A., Wang, E.T., Disney, M. (2020). Small-molecule targeted recruitment of a nuclease to cleave an oncogenic RNA in a mouse model of metastatic cancer. Proc Natl Acad Sci USA, 117(5), 2406-2411. [Google Scholar]
- Cromm, P.M., Crews, C.M. (2017). Targeted protein degradation: from chemical biology to drug discovery. Cell Chem Biol, 2017, 1181-1190. [Google Scholar]
- Cromm, P.M., Samarasinghe, K.T.G., Hines, J., Crews, C.M. (2018). Addressing kinase-independent functions of Fak via PROTAC-addressing kinase-independent functions of Fak via PROTAC-mediated degradation. J Am Chem Soc, 140, 17019-17026. [PubMed] [Google Scholar]
- Deng, Y., Wang, C.C., Choy, K.W., Du, Q., Chen, J., Wang, Q., Li, L., Chung, T.K.H., Tang, T. (2014). Therapeutic potentials of gene silencing by RNA interference: principles, challenges, and new strategies. Gene, 538, 217-227. [PubMed] [Google Scholar]
- Ding, Y., Fei, Y., Lu, B. (2020). Emerging new concepts of degrader technologies. Trends Pharmacol Sci, 41(7), 464-474. [PubMed] [Google Scholar]
- Dharmasiri, N., Dharmasiri, S., Estelle, M. (2005). The F-box protein TIR1 is an auxin receptor. Nature, 435, 441-445. [CrossRef] [PubMed] [Google Scholar]
- Drummond, M.L., Henry, A., Li, H., Williams, C.I. (2020). Improved accuracy for modeling PROTAC-mediated ternary complex formation and targeted protein degradation via new in silico methodologies. J Chem Inf Model, 60(10), 5234-5254. [PubMed] [Google Scholar]
- Dybas, J.M., Herrmann, C., Weitzman, M.D. (2018). Ubiquitination and the interface of tumor viruses and DNA damages responses. Curr Opin Virol, 32, 40-47. [PubMed] [Google Scholar]
- Farnaby, W., Koegl, M., Roy J., Whitworth, C., Diers, E., Trainor, N., Zollman, D., Steurer, S., Karolyi-Oezguer, J., Riedmueller, C., Gmaschitz, T., Wachter, J., Dank, C., Galant, M., Sharps, B., Rumpel, K., Traxler E., Gerstberger, T., Schnitzer, R., Petermann, O., Greb, P., Weinstabl, H., Bader, G., Zoephel, A., Weiss-Puxbaum, A., Ehrenhofer-Wolfer, K., Wohrle, S., Boehmelt, G., Rinnenthal, J., Arnhof, H., Wiechens, N., Wu, M.-Y., Owen-Hughes, T., Ettmayer, P., Pearson, M., McConnell, D.B., Ciulli, A. (2019). BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat Chem Biol, 15, 672-680. [PubMed] [Google Scholar]
- Fischer, E.S., Bohm, K., Lydeard, J.R., Yang, H., Stadler, M.B., Cavadini, S., Nagel, J., Serluca, F., Acker, V., Lingaraju, G.M., Tichkule, R.B., Schebesta, M., Forrester, W.C., Schirle, M., Hassiepen, U., Ottl, J., Hild, M., Beckwith, R.E.J., Harper, J.W., Jenkins, J.L., Thomä, N.H. (2014). Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nature, 512, 49-53. [PubMed] [Google Scholar]
- Fisher, S.L., Phillips, A.J. (2018). Targeted protein degradation and the enzymology of degraders. Curr Opin Chem Biol, 44, 47-55. [PubMed] [Google Scholar]
- Flanagan, J.J., Qian, Y., Gough, S.M., Andreoli, M., Bookbinder, M., Cadelina, G., Bradley, J., Rousseau, E., Chandler, J., Willard, R., Pizzano, J., Crews, C.M., Crew, A.P., Taylor, I., Houston, J. (2018). ARV-471, an oral estrogen receptor PROTAC protein degrader for breast cancer. SABCS, San Antonio, Texas, USA, 2018, December 4–8. [Google Scholar]
- Gadd, M.S., Testa, A., Lucas, X., Chan, K.-H., Chen, W., Lamont, D.J., Zengerie, M., Ciulli, A. (2017). Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol, 13, 514-521. [PubMed] [Google Scholar]
- Gandhi, A.K, Kang, J., Havens, C.G., Conklin, T., Ning, Y., Wu, L., Ito, T., Ando, H., Waldman, M.F., Thakurta, A., Klippel, A., Handa, H., Daniel, T.O., Schafer, P.H., Chopra, R. (2014). Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN). Br J Haematol, 164(6), 811-821. [PubMed] [Google Scholar]
- Ghidini, A., Clery, A., Halloy, F., Allain, F.H.T., Hall, J. (2021). RNA-PROTACs: degraders of RNA-binding proteins. Angew Chem Int Ed, 60(6), 3163-3169. [Google Scholar]
- Gordon, D.E., Jang, G.M., et al. (125 authors). (2020). A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature, 583, 459-468. [PubMed] [Google Scholar]
- Haniff, H.S., Tong, Y., Liu, X., Chen, J.L., Suresh, B.M., Andrews, R.J., Peterson, J.M., O’Leary, C.A., Benhamou, R.I., Moss, W.N., Disney, M.D. (2020). Targeting the SARS-CoV‑2 RNA genome with small molecule binders and ribonuclease targeting chimera (RIBOTAC) degraders. ACS Cent Sci, 6, 1713-1721. [PubMed] [Google Scholar]
- He, S., Ma, J., Fang, Y., Liu, Y, Wu, S., Dong, G., Wang, W., Sheng, C. (2020). Homo-PROTAC mediated suicide of MDM2 to treat non-small cell lung cancer. Acta Pharm Sinica B. DOI: 10.1016/j.apsb.2020.11.022. [Google Scholar]
- Henning, N.J., Boike, L., Jessica N., Spradlin, J.N., Ward, C.C., Belcher, B., Brittain, S.M., Hesse, M., Dovala, D., McGregor, L., McKenna, J., Tallico, J.A., Schirle, M., Nomura, D.K. (2021). Deubiquitinase-targeting chimeras for targeted protein stabilization. bioRxiv preprint. DOI: 10.1101/2021.04.30.441959. [Google Scholar]
- Itoh, Y., Ishikawa, M., Naito, M., Hashimoto, Y. (2010). Protein knockdown using methyl bestatin–ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J Am Chem Soc, 132, 5820-5826. [PubMed] [Google Scholar]
- Kargbo, R.B. (2020). SMARCA2/4 PROTAC for targeted protein degradation and Cr therapy. ACS Med Chem Lett, 11, 1797-1798. [PubMed] [Google Scholar]
- Karim, M., Biquand, M., Declercq, M., Jacob, Y., van der Werf, S., Demeret, C. (2020). Nonproteolytic K29-linked ubiquitination of the PB2 replication protein of influenza A viruses by proviral Cullin 4-based E3 ligases. mBio, 11(2), e00305-20. [PubMed] [Google Scholar]
- Kenten, J.H., Roberts, S.F., Lebowitz, M.S. (2000). Controlling protein levels in eukaryotic organisms using novel compounds comprising a ubiquitination recognition element and a protein binding element. WO2000047220A1. [Google Scholar]
- Kim, J., Kim, H., Park, S.B. (2014). Privileged structures: efficient chemical ′′navigators′′ toward unexplored biologically relevant chemical spaces. J Am Chem Soc, 136, 14629-14638. [PubMed] [Google Scholar]
- Konstantinidou, M., Li, J., Zhang, B., Wang, Z., Shaabani, S., Ter Brake, F., Essa, K., Dömling, A. (2019). PROTACs- a game-changing technology. Expert Opin Drug Discov, 14(12), 1255-1268 [PubMed] [Google Scholar]
- Krönke, J., Udesshi, N.D., Narla, A., Grauman, P., Hurst, S.N., McConkey, M., Tanya Svinkina, T., Heckl, D., Comer, E., Li, X., Ciarlo, C., Hartman, E., Munshi, N., Schenone, M., Schreiber, S.L., Carr, S.A., Ebert B.L. (2014). Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science, 343(6168), 301-305. [PubMed] [Google Scholar]
- Lai, A.C., Crews, C.M. (2017). Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov, 16(2), 101-114. [PubMed] [Google Scholar]
- Lebraud, H., Wright, D.J.J., Johnson, C.N., Heightman, T.D. (2016). Protein degradation by in-cell self-assembly of proteolysis targeting chimeras. ACS Cent Sci, 2(12), 927-934. [PubMed] [Google Scholar]
- Lipinski, C.A., Lombardo, F., Dominy, B.W., Feeney, P.J. (1997). Experimental and computational approaches to estimate solubility and permeability in drug discovery and permeability in drug discovery and development settings. Adv Drug Delivery Rev, 23, 3-25. [Google Scholar]
- Long, M.J., Gollapalli, D.R., Hedstrom, L. (2012). Inhibitor mediated protein degradation. Chem Biol, 19, 629-637. [PubMed] [Google Scholar]
- Lu, G., Middelton, R.E., Sun, H., Naniong, M., Ott, C.J., Mitsiades, C.S., Wong, K.-K., Bradner, J.E., Kaelin Jr, W.G. (2014). The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science, 343(6168), 305-309. [PubMed] [Google Scholar]
- Luh, L.M., Scheib, U., Juenemann, K., Wortmann, L., Brands, M., Cromm, P.M. (2020). Prey for the proteasome: targeted protein degradation – A medicinal chemist’s perspective.. Angew Chem Int Ed Engl, 59, 15448-15466. [PubMed] [Google Scholar]
- McCoull, W., Cheung, T., Anderson, E., Barton, P., Burgess, J., Byth, K., Cao, Q., Castaldi, M.P., Chen, H., Chiarparin, E., Carbajo, R.J., Code, E., Cowan, S., Davey, P.R., Ferguson, A.D., Fillery, S., Fuller, N.O., Gao, N., Hargreaves, D., Howard, M.R., Hu, J., Kawatkar, A., Kemmitt, P.D., Leo, E., Molina, D.M., O’Connell, N., Petteruti, P., Rasmusson, T., Raubo, P., Rawlins, P.B., Ricchiuto, P., Robb, G.R., Schenone, M., Waring, M.J., Zinda, M., Fawell, S., Wilson, D.M. (2018). Development of a novel B-cell lymphoma 6 (BCL6) PROTAC to provide insight into small molecule targeting of BCL6. ACS Chem Biol, 13, 3131-3141. [PubMed] [Google Scholar]
- Mahon, C., Krogan, N.J., Craig, C.S., Pick, E. (2014). Cullin E3 ligases and the rewiring by viral factors. Biomolecules, 4, 897-930. [PubMed] [Google Scholar]
- Maniaci, C., Hughes, S.J., Testa, A., Wenzhang, C., Lamont, D.J., Rocha, S., Alessi, D.R., Romeo, R., Ciulli, A. (2017). Homo-PROTACs: bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat Commun, 8, 830. [PubMed] [Google Scholar]
- Min, J.H. (2002). Structure of an HIF-1alpha-pVHL complex: hydroxyproline recognition in signaling. Science, 296, 1886-1889. [CrossRef] [PubMed] [Google Scholar]
- Mullard, A. (2019a). Arvinas’s PROTACs Pass First Safety and PK Analysis. Nat Rev Drug Discov, 18(12), 895. DOI: 10.1038/d41573-019-00188-4. [PubMed] [Google Scholar]
- Mullard, A. (2019b). First targeted protein degrader hits the clinic. Nat Rev Drug Discov, 18, 237-239. [Google Scholar]
- Mullard, A. (2021). Targeted protein degraders crowd into the clinic. Nat Rev Drug Discov, 20, 247-250. [PubMed] [Google Scholar]
- Neklesa, T.K., Crews, C.M. (2012). Chemical biology: greasy tags for protein removal. Nature, 487(7407), 308-309. [PubMed] [Google Scholar]
- Neklesa, T.K., Tae, H.S., Schneekloth, A.R., Stulberg, M.J., Corson, T.W., Sundberg, T.B., Raina, K, Holley, S.A., Crews, C.M. (2011). Small-molecule hydrophobic tagging-induced degradation of HaloTag fusion proteins. Nat Chem Biol, 7(8), 538-543. [PubMed] [Google Scholar]
- Neklesa, T., Snyder, L.B., Willard, R.R., Vitale, N., Pizzano, J., Gordon, D.A., Bookbinder, M., Macaluso, J., Dong, H., Ferraro, C., Wang, G., Wang, J., Crews, C.M., Houston, J., Crew, A.P., Taylor, I. (2019). ARV-110: an oral androgen receptor PROTAC degrader for prostate cancer. J Clin Oncol, 37(7), suppl. 259, ASCO-GU: San Francisco, California, USA, February 14–16. [CrossRef] [Google Scholar]
- Nishiguchi, G., Keramatnia, F., Min, J., Chang, Y., Jonchere, B., Das, S., Actis, M., Price, J., Chepyala, D., Young, B., McGowan, B., Slavish, P.J., Mayasundari, A., Jarusiewicz, J.A., Yang, L., Yong, L., Fu, X., Garrett, S.H., Papizan, J.B., Kodali, K., Peng, J., Pruett Miller, S.M., Roussel, M.F., Mullighan, C., Fischer, M., Rankovic, Z. (2021). Identification of potent, selective, and orally bioavailable small-molecule GSPT1/2 degraders from a focused library of cereblon modulators. J Med Chem, 64, 7296-7311. [PubMed] [Google Scholar]
- Nowak, R.P., Jones, L.H. (2021). Target validation using PROTACs: applying the four pillars framework. SLAS Discov, 26(4), 474-483. [PubMed] [Google Scholar]
- Nowak, R.P., DeAngelo, S.L., Buckley, D., He, Z., Donovan, K.A., An, J., Safaee, N., Jedrychowski, M.P., Ponthier, C.M., Ishoey, M., Zhang T., Mancias, J.D., Gray, N.S., Bradner, J.E., Fischer, E.S. (2018). Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat Chem Biol, 14, 706-714. [PubMed] [Google Scholar]
- Nunes, J., McGonagle, G.A., Eden, J., Kiritharan, G., Touzet, M., Lewell, X., Emery, J., Eidam, H., Harling, J.D., Anderson, N.A. (2019). Targeting IRAK4 for degradation with PROTACs. ACS Med Chem Lett, 10, 1081-1085. [PubMed] [Google Scholar]
- Olson, C.M., Jiang, B., Erb, M.A., Liang, Y., Doctor, Z.M., Zhang, Z., Kwiatkowski, N., Boukhali, M., Green, J.L., Haas, W., Nomanbhoy, T., Fischer, E.S., Young, R.A., Bradner, J.E., Winter, G.E., Gray, N.S. (2018). Pharmacological per-turbation of CDK9 using selective CDK9 inhibition or degradation. Nat Chem Biol, 14, 163-170. [PubMed] [Google Scholar]
- Ottis, P., Crews, C.M. (2017). Proteolysis-targeting chimeras. Induced protein degradation as a therapeutic strategy. ACS Chem Biol, 12, 892-898. [PubMed] [Google Scholar]
- Petterson, M., Crews, C.M. (2019). PROteolysis TArgeting Chimeras (PROTACs) − Past, present and future. Drug Discov Today: Technologies, 31, 15-27. [Google Scholar]
- Pineda, C.T., Ramanathan, S., Fon Tacer, K., Weon, J.L., Potts, M.B., Ou, Y.H., White M.A., Potts, P.R. (2015). Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell, 160(4), 715-728. [PubMed] [Google Scholar]
- Popow, J., Arnhof, H., Bader, G., Berger, H., Ciulli, A., Covini, D., Dank, C., Gmaschitz, T., Greb, P., Karolyi-Ozguer, J., Koegl, M., McConnell, D.B., Pearson, M., Rieger, M., Rinnenthal, J., Roessler, V., Schrenk, A., Spina, M., Steurer, S., Trainor, N., Traxler, E., Wieshofer, C., Zoephel, A., Ettmayer, P. (2019). Highly selective PTK2 proteolysis targeting chimeras to probe focal adhesion kinase scaffolding functions. J Med Chem, 62, 2508-2520. [PubMed] [Google Scholar]
- Reboud-Ravaux, M. (2021). Le protéasome, la seconde vie d’une cible thérapeutique validée : aspects structuraux et nouveaux inhibiteurs. Biologie Aujourd’hui, 215. [Google Scholar]
- Riching, K.M., Mahan, S., Corona, C.R., McDougall, M., Vasta, J.D., Robers, M.B., Urh, M., Daniels, D.L. (2018). Quantitative live-cell kinetic degradation and mechanistic profiling of PROTAC mode of action. ACS Chem Biol, 13(9), 2758-2770. [PubMed] [Google Scholar]
- Roy, R.D., Rosenmund, C., Stefan, M.I. (2017). Cooperative binding mitigates the high-dose hook effect. BMC Syst Biol, 11, 74-84. [PubMed] [Google Scholar]
- Roy, M, Bader, G., Diers, E., Farnaby, W., Ciulli, A. (2019). Crystal structure of PROTAC 1 in complex with the bromodomain of human SMARCA2 and pVHL:ElonginC:ElonginB. DOI: 10.2210/pdb6HAY/pdb. [Google Scholar]
- Saenz, D.T., Fiskus, W., Qian, Y., Manshouri, T., Rajapakshe, K., Raina, K., Coleman, K.G., Crew, A.P., Shen, A., Mill, C.P., Sun, B., Qiu, P., Kadia, T.M., Pemmaraju, N., DiNardo, C., Kim, M.S., Nowak, A.J., Coarfa, C., Crews, C.M., Verstovsek, S., Bhalla, K.N. (2017). Novel BET protein proteolysis-targeting chimera exerts superior lethal activity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm secondary (s) AML cells. Leukemia, 31, 1951–1961. [PubMed] [Google Scholar]
- Salami, J., Crews, C.M. (2017). Waste disposal-an attractive strategy for cancer therapy. Science, 355, 1163-1167. [PubMed] [Google Scholar]
- Salami, J., Alabi, S., Willard, R.R., Vitale, N.J., Wang, J., Dong, H., Jin, M., McDonnell, D.P., Crew, A.P., Neklesa, T.K., Crews, C.M. (2018). Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun Biol, 1, 100, 1-9. [PubMed] [Google Scholar]
- Sakamoto, K.M., Kim, K.B., Kumagai, A., Mercurio, F., Crews, C.M., Deshaies, R.J. (2001). Protacs: chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc Natl Acad Sci USA, 98, 8554-8559. [Google Scholar]
- Sakamoto, K.M., Kim, K.B., Verma, R., Ransick, A., Stein, B., Crews, C.M, Deshaies, R.J. (2003). Development of protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteomics, 2, 1350-1358. [PubMed] [Google Scholar]
- Schapira, M., Calabrese, M.F., Bullock, A.N., Crews, C.M. (2019). Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov, 18, 949-963. [PubMed] [Google Scholar]
- Schiedel, M., Herp, D., Hammelmann, S., Swyter, S., Lehotzky, A., Robaa, D., Ola, J., Ovadi, J., Sippl, W., Jung, M. (2018). Chemically induced degradation of sirtuin 2 (Sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals). J Med Chem, 61, 482-491. [PubMed] [Google Scholar]
- Schiemer, J., Horst, R., Meng, Y., Montgomery, J.I., Xu, Y., Feng, X., Borzilleri, K., Uccello, D.P., Leverett, C., Brown, S., Che, Y., Brown, M.F., Hayward, M.M., Gilbert, A.M., Noe, M.C., Calabrese, M.F. (2021). Snapshots and ensembles of BTK and cIAP1 protein degrader ternary complexes. Nat Chem Biol, 17, 152-160. [PubMed] [Google Scholar]
- Schneekloth, A.R., Pucheault, M., Tae, H.S., Crews, C.M. (2008). Targeted intracellular protein degradation induced by a small molecule: en route to chemical proteomics. Bioorg Med Chem Lett, 18, 5904-5908. [PubMed] [Google Scholar]
- Sheard, L.B., Tan, X., Mao, H., Withers, J., Ben-Nissan, G., Hinds, T.R., Kobayashi, Y., Hsu, F.-F., Sharon, M., Browse, J., He, S.Y., Rizo, J., Howe, G.A., Zheng, N. (2010). Jasmonate perception by inositol phosphate-potentiated COI1-JAZ co-receptor. Nature, 468(7322), 400-405. [PubMed] [Google Scholar]
- Shi, Y., Long, M.J.C., Rosenberg, M.M., Li, S., Kobjack, A., Lessans, P., Coffey, R.T., Hedstrom, L. (2016). Boc3Arg-linked ligands induce degradation by localizing target proteins to the 20S proteasome. ACS Chem Biol, 11 (12), 3328-3337. [PubMed] [Google Scholar]
- Silva, M.C., Ferguson, F.M., Cai, Q., Donovan, K.A., Nandi, G., Patnaik, D., Zhang, T., Huang, H.-T., Lucent, D. (2019). Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. eLife., 8, e45457. [PubMed] [Google Scholar]
- Soares, P., Gadd, M.S., Frost, J., Galdeano, C., Ellis, L., Epemolu, O., Rocha, S., Read, K.D., Ciulli, A. (2018). Group-based optimization of potent and cell-active inhibitors of the von Hippel-Lindau (VHL) E3 ubiquitin ligase: structure-activity relationships leading to the chemical probe (2S, 4R)-1-((S)-2-(1-cyanocyclopropane carboxamido)-3, 3-dimethyl butanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (VH298). J Med Chem, 61(2), 599-618 [PubMed] [Google Scholar]
- Suh, J., Yoo, S.H., Kim, M.G., Jeong, K., Ahn, J.Y., Kim, M.S., Chae, P.S., Lee, T.Y., Lee, J., Jang, Y.A., Ko, E.H. (2007). Cleavage agents for soluble oligomers of amyloid beta peptides. Angew Chem Int Ed Engl, 46(37), 7064-7067. [PubMed] [Google Scholar]
- Sun, Y., Ding, N., Song, Y., Yang, Z., Liu, W., Zhu, J., Rao, Y. (2019). Degradation of Bruton’s tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas. Leukemia, 33, 2105-2110. [PubMed] [Google Scholar]
- Sun, X., Wang, J., Yao, X., Zheng, W., Mao, Y., Lan, T., Wang, L., Sun, Y., Zhang, X., Zhao, Q., Zhao, J., Xiao, R.P., Zhang, X., Ji, G., Rao, Y. (2019). A chemical approach for global protein knockdown from mice to non-human primates. Cell Discov, 5, 10. DOI: 10.1038/s41421-018-0079-1. [CrossRef] [PubMed] [Google Scholar]
- Taillandier, D. (2021). Contrôle des voies métaboliques par les enzymes E3 ligases : une opportunité de ciblage thérapeutique. Biologie Aujourd’hui, 215. [Google Scholar]
- Takahashi, D., Moriyama, J., Nakamura, T., Miki, E., Takahashi, E., Sato, A., Akaike, T., Itto-Nakama, K., Arimoto, H. (2019). AUTACs: cargo-specific degraders using selective autophagy. Mol Cell, 76, 797-810. [CrossRef] [PubMed] [Google Scholar]
- Tan, X., Calderon-Villalobos, A.C., Sharon, M., Zheng, C., Robinson, C.V., Mark, E., Zheng, N. (2007). Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature, 446, 640-645. [CrossRef] [PubMed] [Google Scholar]
- Testa, A., Lucas, X., Castro, G.V., Chan, K.-H., Wright, J.E., Runcie, A.C., Gadd, M.S., Harrison, T.A., Ko, E.-J., Fletcher, D., Ciulli, A. (2018). 3- Fluoro-4-hydroxyprolines: synthesis, conformational analysis, and stereoselective recognition by the VHL E3 ubiquitin ligase for targeted protein degradation. J Am Chem Soc, 140, 9299-9313. [CrossRef] [PubMed] [Google Scholar]
- Testa, A., Hughes, S.J., Lucas, X., Wright, J.E., Ciulli, A. (2020). Structure-based design of a macrocyclic PROTAC. Angew Chem Int Ed, 59, 1727-1734. [CrossRef] [Google Scholar]
- Tomoshige, S., Hashimoto, Y., Ishikawa, M. (2016). Efficient protein knockdown of HaloTag-fused proteins using hybrid molecules consisting of IAP antagonist and HaloTag ligand. Bioorg Med Chem, 24(14), 3144-3148. [CrossRef] [PubMed] [Google Scholar]
- Toure, M., Crews, C.M. (2016). Small-molecule PROTACs. New approaches to protein degradation. Angew Chem Int Ed, 55, 1966–1973. [CrossRef] [Google Scholar]
- Tutland, T., Ethell, B., Kosaka, T., Blasco, F., Zang, R.X., Jain, M., Gould, T., Hoffmaster, K. (2014). Implementation of pharmacokinetic and pharmacodynamic strategies in early research phases of drug discovery and development at Novartis Institute of Biomedical Research. Front Pharmacol, 5, 174. DOI: 10.3389/fphar.2014.00174. [PubMed] [Google Scholar]
- Valeur, E., Guéret, S.M., Adhihou, H., Gopalakrishnan, R., Lemurell, M., Waldmann, H., Grossmann, T.N., Plowright, A.T. (2017). New modalities for challenging targets in drug discovery. Angew Chem Int Ed Engl, 21(35), 10294-10323. [CrossRef] [Google Scholar]
- Walters, W.P. (2012). Going further than Lipinski’s rule in drug design. Exp Opin Drug Discovery, 7, 99-107. [CrossRef] [Google Scholar]
- Wang, Y., Jiang, X., Feng, F., Liu, W., Sun, H. (2020). Degradation of proteins by PROTACs and other strategies. Act Pharm Sin B, 10(2), 207-238. [CrossRef] [Google Scholar]
- Watt, G.F, Scoot-Stevens, P., Gaohua, L. (2019). Targeted protein degradation in vivo with proteolysis targeting chimeras: current status and future considerations. Drug Disc Today, 31, 69-80. [CrossRef] [Google Scholar]
- Winter, G.E., Buckley, D.L., Paulk, J., Roberts, J.M., Souza, A., Dhe-Paganon, S., Bardner, J.E. (2015). Phthalimide conjugation as a strategy for in vivo target protein degradation. Science, 348, 1376-1381. [CrossRef] [PubMed] [Google Scholar]
- Wu, P., Nielsen, T.E., Clausen, M.H. (2015). FDA-approved small- molecule kinase inhibitors. Trends Pharmacol Sci, 36, 422-439. [CrossRef] [PubMed] [Google Scholar]
- Wu, H.Q., Baker, D., Ovaa, H. (2020). Small molecules that target the ubiquitin system. Biochem Soc Trans, 48, 479-497. [CrossRef] [PubMed] [Google Scholar]
- Zaidman, D., Prilusky, J., London, N. (2020). PRosettaC: Rosetta based modeling of PROTAC mediated ternary complexes. J Chem Inf Model, 60, 4894-4903. [CrossRef] [PubMed] [Google Scholar]
- Zheng, N., Shabek, N. (2017). Ubiquitin ligases: structure, function, and regulation. Annu Rev Biochem, 86, 129-157. [PubMed] [Google Scholar]
- Zorba, A., Nguyen, C., Xu, Y., Starr, J., Borzilleri, K., Smith, J., Zhu, H., Farley, K.A., Ding, W., Schiemer, J., Feng, X., Chang, J.S., Uccello, D.P., Young, J.A., Garcia-Irrizary, C.N., Czabaniuk, L., Schuff, B., Oliver, R., Montgomery, J., Hayward, M.M., Coe, J., Chen, J., Niosi, M., Luthra, S., Shah, J.C., El-Kattan, A., Qiu, X., West, G.M., Noe, M.C., Shanmugasundaram, V., Gilbert, A.M., Brown, M.F., Calabrese, M.F. (2018). Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc Natl Acad Sci USA, 115, E7285-E7292. [CrossRef] [Google Scholar]
- Zou, Y., Ma, D., Wang, Y. (2019). The PROTAC technology in drug development. Cell Biochem Funct, 37, 21-30. [CrossRef] [PubMed] [Google Scholar]
Citation de l’article : Reboud-Ravaux, M. (2021). Dégradation induite des protéines par des molécules PROTAC et stratégies apparentées : développements à visée thérapeutique. Biologie Aujourd’hui, 215, 25-43
Liste des tableaux
Exemples de E3 ligases utilisées couramment dans la production de PROTAC. Leurs ligands sont indiqués ainsi le type de protéines qui ont été ciblées.
PROTAC et colles moléculaires en préclinique et en clinique (adapté de Chamberlain & Hamann, 2019).
Liste des figures
|
Figure 1 Contrôle de l’action délétère d’une protéine impliquée dans une pathologie. A. Les deux modèles pharmacologiques classiques : interaction de la protéine avec un inhibiteur à forte concentration pour contrôler la fonction anormale ; contrôle de l’abondance de la protéine en diminuant son niveau intracellulaire par des technologies basées sur les acides nucléiques ou par des dégradations sélectives induites par des petites molécules. Alors que des petites molécules sont utilisées pour inhiber l’action d’une protéine en bloquant son site actif, les petites molécules PROTAC cooptent la machinerie cellulaire pour dégrader complètement la protéine cible. B. Résumé des techniques permettant le contrôle de l’abondance d’une protéine. C. Nombre de publications portant sur la technologie PROTAC au cours des dernières années. |
| Dans le texte | |
|
Figure 2 La compétition cinétique entre vie et mort des protéines gérée par l’UPS. Les différentes possibilités d’inhibition par de petites molécules exogènes de la dégradation de protéines sont indiquées : inhibition des enzymes E1, E2 et E3 ; inhibition de l’interaction E2-E3 ; inhibition du recrutement du substrat protéique par E3 ; inhibition des différentes activités catalytiques du protéasome ; inhibition des activités dé-ubiquitinase et ATPasique du protéasome 26S ; inhibition de la fixation de la chaîne de poly-ubiquitine par les sous-unités Rpn1, 10 et 13 de la particule régulatrice 19S. La protéine poly-ubiquitinylée liée au protéasome 26S peut être soit dé-ubiquitinylée (elle survit), soit dégradée en peptides à la suite d’une activation allostérique. |
| Dans le texte | |
|
Figure 3 Représentation schématique de la dégradation ciblée d’une protéine induite par une molécule PROTAC dans un contexte cellulaire. A. Schématisation de l’ensemble du processus. La molécule PROTAC bifonctionnelle comporte un élément capable de recruter une ligase E3 et un élément recrutant la protéine à dégrader reliés par un espaceur. En se fixant sur ces deux cibles, la molécule PROTAC induit la formation d’un complexe protéine-PROTAC-E2-E3 permettant à la ligase d’ubiquitinyler la protéine d’intérêt sur un résidu lysine de surface. Quand la protéine d’intérêt est suffisamment ubiquitinylée, elle peut être dégradée par le protéasome 26S. À noter, le recyclage du PROTAC en accord avec son action catalytique. B. Schématisation de la taille relative d’une molécule PROTAC par rapport à celle d’une E3 et d’une protéine-cible. |
| Dans le texte | |
|
Figure 4 Exemples de molécules PROTAC avec indication de la nature de la protéine-substrat et de la ligase E3 ciblée. |
| Dans le texte | |
|
Figure 5 Composition des PROTAC et schématisation de leur conception. A. Recrutement par un PROTAC original de différentes protéines-cibles impliquées dans des pathologies via différentes ubiquitine ligases E3. Les protéines sont alors ubiquitinylées et dirigées vers le protéasome 26S qui catalysera leur dégradation. Les molécules PROTAC sont constituées d’un ligand spécifique de la protéine à cibler, d’un espaceur de diverse nature et d’un ligand spécifique d’une E3 donnée. B. Élaboration rationnelle des PROTAC pouvant se lier à différentes E3 (VHL, MDM2, CRBN et ClAP1) tout en utilisant différents ligands de protéines spécifiques via des espaceurs de structure variable. |
| Dans le texte | |
|
Figure 6 Formation en fonction de la concentration de PROTAC d’un complexe ternaire E3-PROTAC-protéine ou de complexes binaires (effet « hameçon »). Une coopération positive entre protéine et E3 (α > 1) sera favorable à l’ubiquitinylation de la protéine contrairement à une coopération négative (α < 1). |
| Dans le texte | |
|
Figure 7 Stratégie générale de synthèse intracellulaire de PROTAC en utilisant la chimie Clic par cycloaddition alcyne-azide avec catalyse par Cu(I) (A) ou cycloaddition de Diels-Alder entre une tétrazine et un alcène contraint (B). Ces réactions sont très sélectives et n’interfèrent pas avec les processus biochimiques. Les constantes de vitesse des réactions chimiques sont de l’ordre de 102 M−1.s−1(A) et de 104 M−1.s−1(B). Les chimères ainsi synthétisées à l’intérieur de la cellule sont appelées CLIPTAC et ont été utilisées pour les E3 VHL (A) et CRBN (B). Les ligands modifiés devant pénétrer dans la cellule ont un poids moléculaire très inférieur à celui du PROTAC. |
| Dans le texte | |
|
Figure 8 Comparaison entre les stratégies de dégradation induite par les PROTAC et par marquage hydrophobe (colle moléculaire) basé ou non sur HaloTag. A. Schématisation de l’interaction entre une protéine-cible et une E3 (CRBN) médiée par une colle moléculaire (a) ou un PROTAC (b). Le poids moléculaire de la colle moléculaire est inférieur à celui du PROTAC. B. Dégradation d’une protéine cible induite par une molécule HyT conduisant à un repliement incorrect. La molécule HyT provoque la déstabilisation de la conformation de la protéine avec complexation à HSP70 (modèle 1) ou une complexation directe avec HSP70 (modèle 2) conduisant dans les deux cas à une dégradation par le protéasome 26S. Le fonctionnement physiologique pour une protéine mal conformée est indiqué pour comparaison. C. Élaboration d’une molécule HyT ciblant la protéine Tau. Un motif hydrophobe adamantyle ou Boc3Arg est lié par covalence à un peptide reconnaissant spécifiquement Tau, le motif poly-arginine favorisant la pénétration cellulaire. La structure d’un PROTAC dégradant la protéine Tau est indiquée pour comparaison. D. Marquage hydrophobe sur une protéine de fusion HaloTag. La protéine de fusion « protéine d’intérêt-HaloTag » est reconnue par une molécule bifonctionnelle. L’état partiel de dénaturation qu’elle induit provoque la dégradation par le protéasome. |
| Dans le texte | |
|
Figure 9 Homo-PROTAC et RIBOTAC. A. Structure d’un homo-PROTAC ciblant la E3 ligase MDM2 (1) et d’un homo-PROTAC ciblant la E3 ligase VHL (2). Ces Homo-PROTAC ont été conçus pour provoquer l’auto-dégradation par le protéasome de chacune de ces ligases. B. Schématisation du mode d’action d’un RIBOTAC. |
| Dans le texte | |
Les statistiques affichées correspondent au cumul d'une part des vues des résumés de l'article et d'autre part des vues et téléchargements de l'article plein-texte (PDF, Full-HTML, ePub... selon les formats disponibles) sur la platefome Vision4Press.
Les statistiques sont disponibles avec un délai de 48 à 96 heures et sont mises à jour quotidiennement en semaine.
Le chargement des statistiques peut être long.